
An Analysis Of FDA FY2017 Drug GMP Warning Letters
By Barbara Unger, Unger Consulting Inc.
 This article presents a detailed summary of the drug GMP warning letters issued in FY2017, as well as a comparison of trends since fiscal year 2013. A comprehensive GMP intelligence program includes evaluation of health authority enforcement actions, including FDA Form 483s, warning letters, seizures, recalls, and consent decree agreements. This allows manufacturers and sponsors to identify new trends and the focus of FDA inspectors and act to address or justify similar situations at their own firms. Section 4.2 of ICH Q10, Pharmaceutical Quality System, specifies the “Monitoring of Internal and External Factors Impacting the Pharmaceutical Quality System” including “Emerging regulations, guidance and quality issues…”. Enforcement actions should be monitored as a component of quality issues mentioned in the guidance. Although the FDA is more transparent than other health authorities regarding enforcement actions, readers are encouraged to follow information published by Health Canada (Drug & health product inspections and Inspection Tracker: Drug Manufacturing Establishments), Eudra GMDP reports of noncompliance, and WHO Notices of Concern.
This article presents a detailed summary of the drug GMP warning letters issued in FY2017, as well as a comparison of trends since fiscal year 2013. A comprehensive GMP intelligence program includes evaluation of health authority enforcement actions, including FDA Form 483s, warning letters, seizures, recalls, and consent decree agreements. This allows manufacturers and sponsors to identify new trends and the focus of FDA inspectors and act to address or justify similar situations at their own firms. Section 4.2 of ICH Q10, Pharmaceutical Quality System, specifies the “Monitoring of Internal and External Factors Impacting the Pharmaceutical Quality System” including “Emerging regulations, guidance and quality issues…”. Enforcement actions should be monitored as a component of quality issues mentioned in the guidance. Although the FDA is more transparent than other health authorities regarding enforcement actions, readers are encouraged to follow information published by Health Canada (Drug & health product inspections and Inspection Tracker: Drug Manufacturing Establishments), Eudra GMDP reports of noncompliance, and WHO Notices of Concern.
The data presented herein for FY2017, ending Sept. 31, 2017, is based on drug GMP warning letters posted by the FDA no later than Oct. 25, 2017. A complete list of FY2013 – FY2017 drug GMP warning letters is provided here.
The term “compounding pharmacy” used herein includes outsourcing facilities and is considered as a category separate from drug manufacturers based on their legal foundation. These sites are all located in the United States, but they are not combined with data from U.S. drug manufacturing sites. Outsourcing facilities were established by the FDA as entities under an amendment to the Food Drug & Cosmetic Act in November of 2013. Compounding pharmacies are governed by state law.
The narrative, tables, and graphics address three broad areas:
- Type of manufacture (API, dosage form, API and dosage form, compounding pharmacy/outsourcing facility), country associated with the warning letter and whether and when an import alert was put in place, and the intervals between inspection and imposition of the import alert.
- Particular targets of warning letters issued this year, including combination products, homeopathic firms, cell and tissue firms, and contracted operations.
- Interval between the inspection and enforcement actions, including issuance of a warning letter or import alert.
Following are brief summaries of noteworthy findings and trends, followed by a more detailed analysis in subsequent sections:
- The number of drug GMP warning letters continues to increase over the previous year from 42 in FY2015 to 102 in FY2016 to 114 in FY2017. (Table 1 and Figure 1)
- The compounding pharmacy/outsourcing facility segment continues to receive disproportionate enforcement attention from the FDA. We may, though, have turned the corner on these warning letters compared with the past few years in both absolute number and in percentage of the total number of warning letters. (Table 1 and Figure 1)
- The number of FY2017 warning letters issued to firms that are either API manufacturers or both API manufacturers and dosage form manufacturers remains essentially the same as in FY2016. (Table 1 and Figure 2)
- The most significant change this year in the type of sites that received a warning letter is a doubling in the number of warning letters issued to drug product manufacturing sites. FY2016 saw 23, while FY2017 saw 46. (Table 1, Figure 2)
- Excluding the compounding pharmacies and outsourcing facilities, the FDA continues to focus its enforcement actions outside the U.S. Two and a half times as many warning letters were issued to firms outside the U.S. compared with those issued to domestic firms. (Tables 1 and 2, Figures 1, 3, 3A)
- At least 11 warning letters were associated with contract operations, issued to either/both the contract site and its customers. (Table 1A)
- Import alerts were associated with 20 of the 49 warning letters issued to sites outside the U.S. in FY2017. Firms in China and India that received warning letters were the subject of 16 of the 20 import alerts associated with warning letters. (Table 3)
- The percentage of warning letters that cite deficiencies in data governance/data integrity decreased slightly from 81 percent to 67 percent for firms outside the U.S. Those issued to U.S. firms decreased from 73 percent in FY2016 to 60 percent in FY2017. Data integrity deficiencies are cited in 65 percent of all warning letters, excluding those issued to compounding pharmacies (Table 4 and Figure 4).
- The interval between inspection and issuance of warning letters is decreasing significantly for all categories, though the most dramatic decrease has been for warning letters issued to sites outside the U.S. (Table 5 and Figures 5 and 6)
Warning Letter Data
Table 1 shows that drug GMP warning letters more than doubled from FY2015 to FY2016 and increased by approximately 12 percent from FY2016 to FY2017. Table 1 also shows that while the FDA continues an intense focus on compounding pharmacies, the warning letters to these entities appear to have turned a corner and have decreased significantly. For the first time since FY2013, the percentage of the total drug warning letters issued to these firms is now 39 percent, where for the past three years it has been above 50 percent. The FDA’s legal authority over these entities was clarified in the Drug Quality and Security Act (DQSA), signed in November of 2013, and explains the apparent explosion of enforcement action in this area beginning in FY2014. It will be interesting to watch and see whether FY2018 continues this trend.
Warning letters issued to API sites increased since FY2013, and the number is the same this year as it was last year. The most significant change is the doubling in the number of warning letters issued to drug product sites, doubling from 23 in FY2016 to 46 in FY2017.
Within the category of drug product manufacturers are three homeopathic drug manufacturers and two cellular or tissue manufacturing sites. We’ll watch how this develops in FY2018. The FDA has stated it will evaluate the entire area of homeopathic drugs and its regulation.
In addition, the FDA is actively supporting the development of stem cell treatments and other types of regenerative therapies — and attempting to differentiate them from the hundreds of stem cell clinics that make broad and unfounded claims of safety and efficacy. The FDA has issued several warning letters in the past year to clinics that deliver services and products of this type, determining they posed an immediate public safety risk.
Also, the agency recently published a 24-page final guidance titled Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use. This guidance addresses the FDA compliance policy for these product types, stating that manufacturers will have 36 months to prepare either an IND or marketing application. During that time, FDA will “generally” exercise enforcement discretion for products that do not meet all criteria identified in 21 CFR 1271.10(a) if the human cell, tissue, or cellular and tissue-based product (HCT/P) is “intended for autologous use and its use does not raise reported safety concerns or potential significant safety concerns.”
Another interesting topic in 2017’s warning letters is, in my analysis, the second warning letter to cite deficiencies in manufacture of combination products. Meridian Medical Technologies, Inc., a Pfizer Company, received a warning letter for the EpiPen it manufactures for Mylan. This follows Amgen’s 2014 warning letter, which was the first to cite the new 21 CFR 4 regulation governing these products. Certainly, other warning letters have been issued to manufacturers of prefilled syringes and pen injectors, but only these two cite the combination product regulations.
FY2017 warning letters also had a focus on contracted activities. At least 11 warning letters mention contract activities. Sixteen percent of warning letters issued to firms that were not compounding pharmacies were directly associated with contract manufacturing or testing. In two instances shown in Table 1A, warning letters were issued to both the contractors and the sponsors of the products in question. In two other instances the sponsor was identified but it did not receive warning letters for the same problem. In most cases, the sponsors were not identified when the contract manufacturer or contract testing laboratory received a warning letter, so it is not possible to determine whether they also received a warning letter.
And finally, while not addressed in a table or figure, the FDA continued to cite failures in aseptic processing of sterile drug products and deficiencies in process validation. Specifically, the FDA cited the failure to implement an ongoing process monitoring program to ensure that the manufacturing process remains in a state of control. This is applicable to all types of drug manufacture, oral and topical dosage forms, as well as sterile injectables.
Table 1: Drug GMP Warning Letters
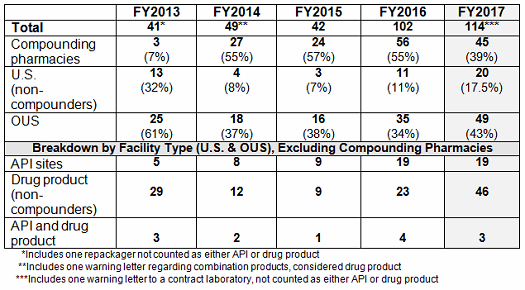
Table 1A: Contract Manufacturers, Sponsors, and Warning Letters
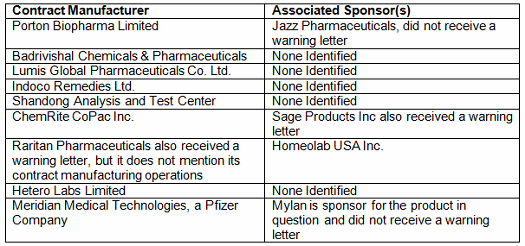
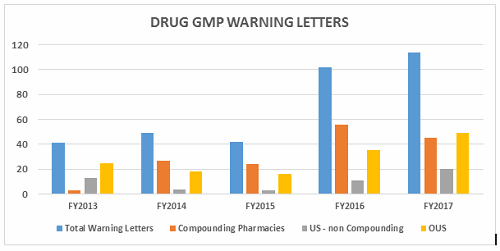
Figure 1
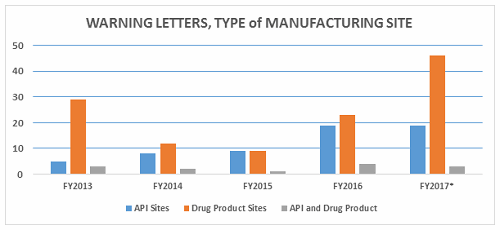
Figure 2
Table 2 shows the geographic distribution of warning letters issued outside the U.S. European countries are counted together. European area countries over the five fiscal years include: Ireland, Spain, Czech Republic, Italy, Portugal, Denmark, Austria, Netherlands, Germany, Hungary, the U.K., and Switzerland. India received the highest number of warning letters issued to a single country over the five-year period. China received the next highest number of warning letters, followed closely by Europe. For the past two years, China received more warning letters than India, though overall, India comes in first. Figure 3 shows the same information. China, India, and Europe accounted for almost 80 percent of drug GMP warning letters issued outside the U.S. over the five-year period; the rest of the world received the remaining approximately 20 percent. With the Mutual Recognition Agreement between the U.S. and the EU, it will be interesting to see if the number of warning letters to European sites begins to decrease in FY2018. Currently, this agreement includes eight health authorities in the EU.
Table 2: Drug GMP Warning Letters Issued Regarding Sites Outside the U.S.
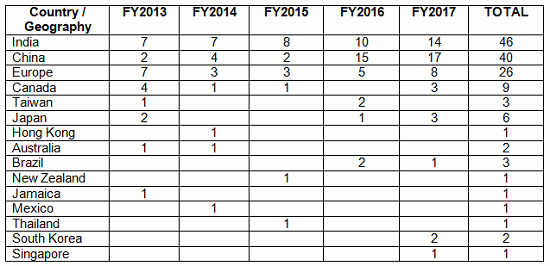
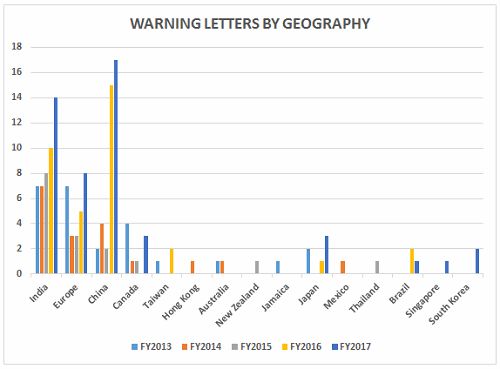
Figure 3
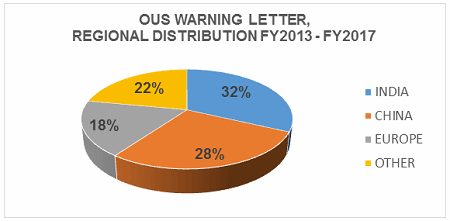
Figure 3A
Import Alerts Associated With Warning Letters
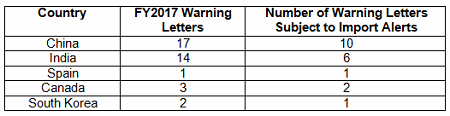
Forty-nine warning letters were issued regarding sites outside the U.S., and 20 of these had associated import alerts for failure to comply with drug GMPs or refusal of an inspection. So not only did these firms have the expense associated with remediation of the warning letter, they are prevented from selling product from these sites in the U.S., excluding FDA-identified medically necessary products. Table 3 shows the distribution of the import alerts associated with warning letters in FY2017. China and India, taken together, account for 80 percent of the import alerts associated with warning letters.
Figure 6 shows that the interval between the inspection and imposition of an import alert decreased in FY2017 compared with FY2016.
Table 3: Import Alerts Associated With FY2017 Warning Letters
Data Integrity Deficiencies In Warning Letters
Table 4 shows the number of warning letters issued both inside and outside the U.S. that included references to data management and data integrity. This group and analysis excludes those warning letters issued to compounding pharmacies. Data integrity deficiencies in warning letters continue to identify the predicate rule(s) to which firms did not adhere. Figure 4 provides a graphic representation of the data. The percentage of warning letters that cite data integrity deficiencies issued to sites in the U.S. is very similar when compared to warning letters issued outside the U.S., though the absolute numbers differ. The numbers and percentages have decreased between FY2016 and FY2017.
Table 4: Data Integrity Deficiencies in Warning Letters, Excluding Compounding Pharmacies
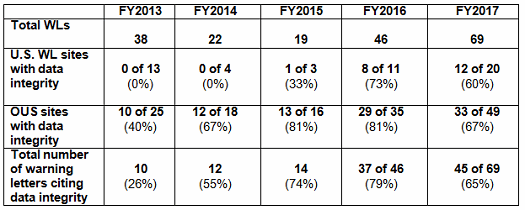
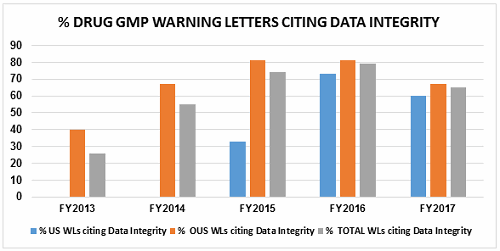
Figure 4
Intervals Between Inspection And Warning Letter
Table 5 and Figures 5 and 6 show the interval between inspection and issuance of a warning letter for the most recent five fiscal years. The data for all three major categories shows a decrease in the time between inspections and warning letter issuance. The most significant change happened for the sites outside the U.S., which saw a decrease from 10.4 months in FY2016 to 8.0 months in FY2017. The time between inspection and warning letter issuance is longest for compounding pharmacies at 12.6 months, though this is about 0.5 month shorter than in FY2016.
For warning letters issued to sites outside the U.S. in FY2017, Figure 6 compares the intervals between inspection and warning letters, and inspection and imposition of an import alert.
Table 5: Months Between Inspection and Warning Letter
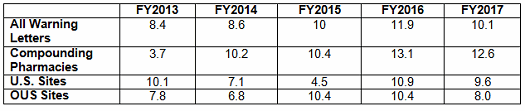
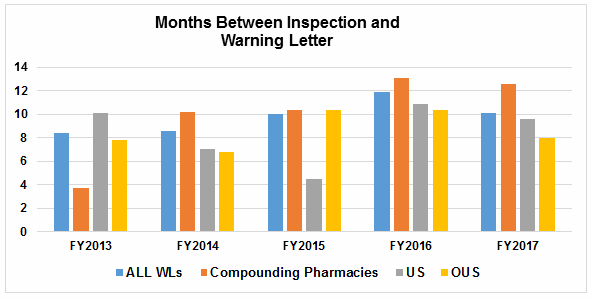
Figure 5
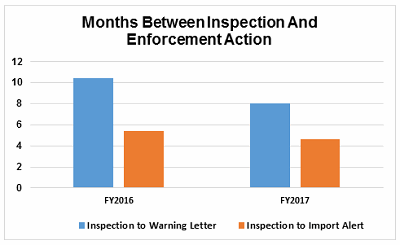
Figure 6
Conclusions
FY2017 saw another year of increase in the number of drug GMP warning letters issued by the FDA, though not as dramatic a difference as between FY2015 and FY2016.
Compounding pharmacies and outsourcing facilities continue to receive disproportionate warning letter enforcement attention. In 2017, however, we may have seen the corner turned because the number and percentage of warning letters issued to firms in this market segment decreased. We will continue to monitor this metric and see whether FY2018 shows a similar decline.
Contract manufacturers and their sponsors were a focus this year, and I would expect that to continue and expand in FY2018. Some sponsors will only take their responsibilities for selection and oversight of contract manufacturers seriously if there is a real economic impact, such as a contract manufacturer that suffers a serious enforcement action that could result in a delay, or even a cease, in manufacture to undertake the necessary remediation.
FY2017 saw several warning letters to manufacturers of homeopathic products, and this seems to be continuing into FY2018. In December, the FDA published a draft guidance that clarifies how it intends to take risk-based enforcement actions against these firms, which represents a clear change from the Compliance Policy Guide, CPG400.400, from 1988. Look for more enforcement, including potentially coordinated actions between the FDA and the Federal Trade Commission (FTC), as the two agencies did in a collection of warning letters to these firms in 2011. Change happens slowly, but the growth of this industry segment is prompting additional attention from the FDA.
China continues to lead in the country receiving warning letters, as it did in FY2016. This year saw the addition of the Republic of (South) Korea to the countries whose companies received warning letters. I would expect this to continue into FY2018 also.
Overall, in FY2018, look for the following trends in enforcement actions:
- Continued focus on compounding pharmacies
- Continued focus on data integrity and data governance
- Continued focus on sites outside the U.S., including China, India, and South Korea. We might see some decline in inspections in some EU countries based on the MRA.
- Contract manufacturers and laboratories and those that contract for their services will see continued attention by the FDA.
- There likely will be increased attention in process validation, particularly ongoing process monitoring, as required in both the FDA and EMA validation guidance.
- Homeopathic product manufacturers and stem cell product manufacturers will see additional enforcement actions based on their overall potential impact on public health and the FDA’s stated focus to improve risk-based enforcement in those areas.
About the Author:
 Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. Barbara is currently the co-lead of the Rx-360 Data Integrity Working Group.
Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. Barbara is currently the co-lead of the Rx-360 Data Integrity Working Group.
Before Amgen, Barbara worked for the consulting firm Don Hill and Associates, providing regulatory and quality services to the pharmaceutical industry, and for Eli Lilly and Company in quality and CMC regulatory affairs positions. She began her career in the pharmaceutical / device industry with Hybritech Inc. and received a bachelor's degree in chemistry from the University of Illinois at Urbana-Champaign. You can contact Barbara at bwunger123@gmail.com.