
Continued Process Verification Of Legacy Products In The Biopharma Industry
By Andre Walker (Andre Walker Consulting), Bert Frohlich (Biopharm Designs), Carly Cox (Pfizer), Julia O’Neill (Tunnell Consulting), Marcus Boyer (BMS), Parag Shah (Genentech Roche), Paul Wong (Bayer), Rob Grassi (Lonza), Robin Payne (BioPhorum) and David Bain (technical writer)

The BioPhorum Operations Group published an industry position paper on continued process verification (CPV) for biologics1 in 2014. It included a detailed example CPV plan for a single biologic molecule based on the process defined in “A-MAb: A Case Study in Bioprocess Development.”2 The paper provided insight into leveraging quality by design (QbD) concepts to enable CPV and a detailed methodology for selecting process parameters from the overall control strategy, defining sampling plans, and analyzing the data that flows through the manufacturing process. BioPhorum members also published a road map for the implementation of CPV3 in 2016.
The FDA,4 European Medicines Agency (EMA),5 and International Conference on Harmonization (ICH)6 have issued guidance stating manufacturers are responsible for identifying sources of variation affecting process performance and product quality. This means that, for all practical purposes, the implementation of systems to ensure continued quality assurance is a GMP requirement. Furthermore, it is expected to extend to existing commercial operations and legacy programs.
This article summarizes how the challenges of implementing a CPV program for a legacy biopharmaceutical product might be overcome, where a product: was licensed before the FDA published its guidance, has a manufacturing history, and may be produced at more than one site and/or where resources for implementing CPV are limited. It draws on the collective experiences of the member companies of BioPhorum Operations Group (BPOG) to suggest appropriate strategies. While targeted at the biopharmaceutical industry, the concepts presented are likely to be applicable to small molecule and emerging therapeutic technologies.
Integrating CPV With Legacy Quality Systems
CPV will likely require new concepts, definitions, and procedures that will have to align with policies and procedures in the existing quality systems. Three specific challenges are conflict over the definition of “validation,” differentiating between the CPV process and existing trending procedures, and integrating the CPV process with the formal exceptions (deviations) system.
Until the FDA’s industry guidance in 2011,4 process validation was a one-time event where batches were produced to provide evidence that the process worked reliably and predictably. The 2011 guidance, however, extends process validation across the product life cycle.
While it may be tempting to rewrite existing procedures so they embrace the new concept, it is likely the old definition of process validation is thoroughly embedded in hundreds of documents and regulatory filings. Modifying these will not deliver the intent of the agency, which is to have a CPV program ensure a state of control. A more reasonable approach is to create one high-level document to codify the vocabulary of the company against the 2011 FDA guidance and act as a key for translating between old and new documents.
A company undoubtedly already trends multiple product attributes and process parameters, and these will be part of the existing quality system. It is important to recognize that these existing procedures focus on licensed specifications and are primarily concerned with safety and efficacy. CPV, on the other hand, is concerned with ensuring the process remains in a state of control. To avoid conflict and confusion between the two, BPOG member companies recommend carefully choosing terms to differentiate between them. For example, CPV charts generate “signals” that are “evaluated,” whereas specification-based systems likely use the terms “trend” and “investigation.”
In rare instances, a CPV signal may be evaluated and found to possibly impact safety and efficacy. In this case, the issue should be escalated into the formal exceptions system, where it will be thoroughly investigated. It is recommended that specific rules for escalation are established, so as not to overburden the exceptions system with signals that don’t deserve extra scrutiny.
Design Space Knowledge And Parameter Selection
Legacy products that were developed years ago were likely licensed, based on a level of process understanding and a control strategy definition that was appropriate at the time but is less sophisticated than current regulatory expectations and industry standards. This means there may be cases where systematic classification of process parameters has not been developed or should be updated as a precursor to defining the CPV program. In other cases, gaps might exist for process data that was not collected on important attributes and parameters, which are needed in the CPV program.
A reasonable strategy that enables relatively rapid and efficient deployment of a compliant CPV program is implementing the new monitoring program in phases, initially focusing on the product qualities of the legacy product that are measured at the time of release. The data should be readily available and already trended against release specification in the annual product review. The CPV program will increase the frequency of data review, establish trending against statistical control limits, and identify the capability of the critical quality attributes (CQAs). It also permits the establishment of a CPV program with a manageable set of product attributes.
A science- and risk-based control strategy is the foundation of an efficient and robust CPV plan. One should be created or updated to define CQAs and critical process parameters (CPPs) that impact CQAs for legacy products by taking advantage of the rich manufacturing data that has been accumulated. Formal risk assessment methods (failure mode and effects analysis, etc.) are the fundamental tools used to classify parameter impact (e.g., critical, key, no impact) and should be employed. Once complete, the control strategy guides the choice of parameters to include in the CPV program.
Legacy Products And Variability Across Multiple Sites
When a legacy product is manufactured at different sites executing the same manufacturing procedures, a chief concern is that a CPV program will highlight process differences between the sites. These differences may stem from facilities that have been built at different times and/or in different companies, design standards and equipment performance that may have changed over time, and facilities that may be operated by different personnel or even by different companies, e.g., CMOs. Performance differences between sites are almost unavoidable, and thus the introduction of a cross-site CPV program may be perceived as only highlighting these differences and offering opportunities for improved consistency and performance.
It is useful to distinguish between a CPV plan, including the list of process parameters and product quality attributes to trend based on the control strategy, and the presentation of data that comes from the different locations. As a guiding principle, for processes being executed in multiple locations the decision to have one or multiple CPV plans is based on the level of difference in process design (or unit operation design); it should not be based on process performance.
Identical processes (those with the same unit operations, CQAs, and CPPs) being executed in different suites, sites, or CMOs are presumably all controlled by the same science- and risk-based control strategy. Thus, the list of control inputs (parameters) and process outputs to be monitored should be the same. Conversely, newer versions of a process (or unit operation) with improved control strategies or different unit operations should be considered a new process with a separate CPV plan. There may be significant overlap in the old and new CPV plans, but the one-to-one relationship between process design and CPV plan should be maintained.
A CPV program configured as noted above will highlight the differences between sites, which should not be cause for concern, as both sites are licensed to manufacture against predetermined release criteria that ensure safety and efficacy. A properly defined CPV program should have no impact on safety and efficacy but should provide early detection of process shifts and drifts and appropriately prompt an escalation of activities that result in greater process understanding and control.
Discovery, Disclosure, And Regulatory Action
It is understandable that some reluctance may exist for implementing systems that will generate new (additional) data about an existing process. What if the data generated is inconsistent with current process understanding and regulatory filings or commitments? What if unexpected levels of variation are discovered? What is the responsibility of the manufacturer in disclosing newly found information?
Perhaps the most significant concern would be that through more thorough data review, the CPV program uncovers an issue that indicates a risk to safety and efficacy of a product that was already released and distributed. This would be a truly rare event. CPV programs are designed to monitor the state of control of the process, to verify the validated state has been maintained, and to generate data that enables process improvement. The CPV trend limits are based on statistically derived control limits, not safety or specification limits. Even if a CPV program establishes that a given parameter has a substandard capability, safety and efficacy are ensured by following the process as it was filed, including all of its in-process controls and release tests.
Furthermore, it is an expectation of health agencies that manufacturing process issues (errors, out-of-specifications, etc.) be identified and investigated and corrective and preventive actions (CAPAs) be initiated to continually improve the process. CPV activities are merely an extension of this continuous-improvement expectation and no more likely to result in significant regulatory concerns than existing process monitoring systems.
To reiterate, the purpose of reviewing historical data is not to re-release past production, nor is it likely this data review will discover information that indicts previous lots. All data from released batches should be within the licensed range, and even if a process is evaluated as having a low process performance indicator (PpK), safety and efficacy were assured by the existing quality systems in place at time of release. Note though that, however unlikely, it is prudent to have an agreed-upon escalation path, should the data review identify an issue with possible patient impact.
Data Integration And Legacy IT Infrastructure
Collection, analysis, and reporting of a significant volume of data is required for the practice of CPV. For older facilities, legacy data acquisition systems and IT infrastructure may not be able to readily access the data required by the CPV protocol. Therefore, a substantial investment may be required to implement CPV.
In addition to cost, there are technical challenges associated with applying a new information system to a manufacturing process that has a lengthy history. Data may have been collected for years in paper batch records, for example, or reside in separate, unconnected systems such as laboratory information management systems (LIMS) or enterprise resource planning (ERP) systems. Monitoring process performance across multiple sites, including CMOs, presents further challenges.
There is no single best solution to address these issues. In each case, there should be a cost-benefit assessment on the value of establishing connected IT systems for the scale and scope of CPV envisioned. Starting with a simple system at one site with the capability of expanding to a comprehensive, multi-site system is generally recommended.
A data hierarchy should be created that maps the various CPV parameters to the primary source of the data. It is also useful to consider two separate challenges: the backward-looking population of a data historian with prior product and process data and the forward-looking system for continuously collecting and analyzing data from the manufacturing process. These can be considered two separate sets of processes, united by the data hierarchy.
Collecting historical information is a one-time event that will be resource-intensive and difficult to automate. This is likely to be achieved most efficiently by hand. IT is a key partner in establishing the infrastructure necessary to automate future data flow and create a sustainable infrastructure for CPV. Data integrity is important in each case, and the effort expended to ensure this should be proportionate to risk.
The Effort And Cost Of Deploying CPV For Legacy Processes
The implementation of a CPV program requires significant commitment from management to supply the necessary human and capital resources. Leadership must also drive the cultural change required to respond to signals from control charts, even as the process is performing within specification limits. Implementations without this level of support may be compliant but certainly will not provide the expected improvement in productivity and efficiency that CPV programs have the potential to create.
If management commitment must be developed, new guidelines from regulatory agencies are a powerful motivator to act on CPV. But developing a compelling business case is paramount to gaining the level of support for an implementation that will provide benefits beyond compliance.
BioPhorum member companies identify three significant benefits of a strong CPV program. The value of each of these should be estimated and used to justify expenditures for the CPV program:
- Sensing process shifts and proactively identifying and fixing underlying issues, which may prevent the loss of future batches
- Sensing process shifts and proactively identifying and fixing underlying issues, thereby reducing deviations/exceptions and associated workload
- Improved process knowledge that can lead to improvement in process control, which will lead to increased process robustness and yield
BioPhorum member companies recommend a phased rollout using a CPV road map.3 Clear CPV program goals in concert with a supportive deployment strategy will focus CPV deployment efforts for maximum benefit, but also provide a unifying message from management to the broader organization. Table 1 highlights three potential goals to address and indicates the basis of a rollout strategy.
Table 1: CPV Rollout Concepts
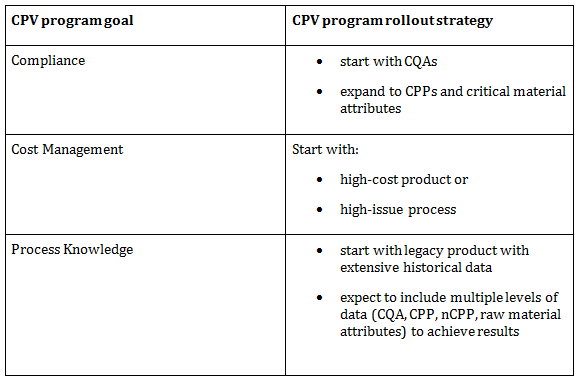
The previous sections discussed the specific challenges that tend to make the implementation of CPV for a legacy program more complicated and, therefore, costlier. However, legacy products have a long manufacturing history where data mining and analysis can result in a very rapid accumulation of process understanding, which is the true value of a CPV program, going beyond compliance to superior performance. This is a key benefit that can be used to justify the additional expenditure.
An extended version of this article is available on the BioPhorum website.
References:
- Continued Process Verification: An Industry Position Paper with Example Plan, BioPhorum Operations Group (2014), https://www.biophorum.com/download/cvp-case-study-interactive-version/
- A-MAb: A Case Study in Bioprocess Development, CMC Biotech Working Group (2009), http://www.ispe.org/pqli/a-mab-case-study-version-2.1
- Boyer M, Gampfer J, Zamamiri A, et al. A Roadmap for the Implementation of Continued Process Verification, PDA J Pharm Sci and Tech 2016, 70 282-292
- Guidance for Industry, Process Validation: General Principles and Practices, US Food and Drug Administration (2011), http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
- Guideline on Process Validation for the Manufacture of Biotechnology-Derived Active Substances and Data to be Provided in the Regulatory Submission, EMA/CHMP/BWP/187338/2014, http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/04/WC500205447.pdf
- ICH Harmonised Tripartite Guideline: Pharmaceutical Quality System Q10, http://www.ich.org/products/guidelines/quality/quality-single/article/pharmaceutical-quality-system.html