
Is Your Sterile Injectable Ready For Changes In Raw Materials?
By Lisa Cherry, Ph.D., Pfizer CentreOne

Whether you work with a CDMO or manufacture your drug at one of your own sites, there will likely be changes in raw materials from time to time. These include modifications in API synthesis, key starter materials, and intermediates. That’s to be expected throughout the lifecycle of a drug. A “best practices” approach to accepting alternate material from a supplier is vital to maintaining drug integrity.
Changes in raw materials may occur for a variety of reasons, such as the adoption of advances in manufacturing processes, availability of “equivalent” less expensive raw materials, or a change in the manufacturing location of a raw material producer. Astute raw-material control personnel manage these changes carefully. They know a seemingly minor change may have a significant impact on the behavior of the raw material in a drug production environment.
The following case studies illustrate a robust materials review process and material control system to assure continuous drug manufacturability, quality, and usability, even when alternate material behaves somewhat differently than the original. Although these case studies pertain to sterile injectables, the principles described and trouble-shooting required apply to small-molecule drugs in general.
Equivalent raw materials
Whenever there’s a pending material change, it is helpful to learn as much as possible about the alternate substance. For instance, is the material going to remain in flake form or change to granule? If it is an API, have any of the process steps changed? Are the residual solvents the same?
Communicating with suppliers throughout the changeover process, and assessing what dimensions are changing, enables a seamless transition to acceptable new material for use in dosage form manufacture.
Case Study One: optimal situation
In Case Study One, we outline an example of what a material control manager would like to see during the qualification of an alternate raw material. The API supplier notified the material manager well in advance of the API change and provided samples with sufficient lead time to evaluate the material and prepare drug product lab batches for comparison purposes.
The lab batches were made with both the currently approved raw material source and the alternate source. The materials team wanted to see that both behaved similarly and that the dosage form manufacturing process would not be affected. Analytically, no detectable differences were found between the APIs or the finished dose form. Both APIs processed similarly in the lab and later at full scale. Stability studies also confirmed that the API change did not affect the drug’s stability. The end result: Both APIs proved comparable.
Case Study Two: precipitate
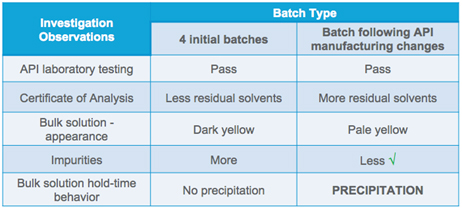
In Case Study Two, we examine what can happen when a seemingly routine API change presents a challenge at the compounding stage of dosage form production. As a matter of routine process, the materials management team tested the incoming API for identification and compared the results with the certificate of analysis (COA). The team moved forward and manufactured a batch and observed no problems throughout the process.
The materials management team took samples during the compounding process to perform lab studies. And that’s where the problem was discovered. One day following the sample acquisition, some precipitate was observed, and the longer the samples sat, more precipitation formed.
An investigation was initiated. In the COA, it was known that lower levels of residual solvents were present with the original material, and relatively higher levels with the newer material. During incoming API release of the original and new API, however, no red flags were raised - both met specifications.
Moreover, a comparison of the bulk solution indicated higher levels of impurities existed with the original material, when compared with the new material. Based on these results, it seemed reasonable that the bulk solution would remain stable. The bulk-solution hold-time behavior of the new material, however, did not match the expectations for stability.
At this point a discussion with the API supplier was initiated to see if going back to the old process was a possibility. It was not. The API producer had already implemented the API process changes on a large scale. The material and production teams were challenged to make the new API work.
Following a comprehensive review, the team found the new API would only solubilize completely at a specific pH value. A pH adjustment was therefore required in order to move the API into solution. The pH adjuster, however, added a counter ion to the API, producing a salt form, which caused the substance to precipitate out of solution.
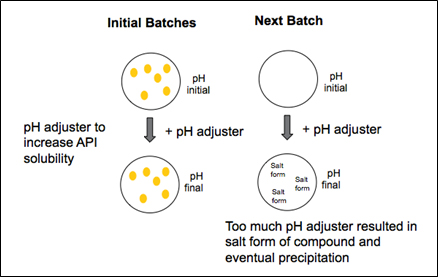
The answer, the team found, was to reduce the amount of pH adjuster. Rather than add a significant amount at the onset to dissolve excipients, heat was used to aid in dissolution. The excipients fully dissolved with the use of heat and no adverse effects were observed in samples held for stability. Upon cooling, the pH was fine-tuned using limited amounts of pH adjuster, as determined through successive iterations of lab batches. Following the pH adjustment, API was added and mixed into the solution.
Gaining control over how much pH adjuster was added to produce the bulk solution, the material manager and production teams fully resolved the precipitate problem. Following resolution of this problem, the manufacturer instituted new protocols for accepting any alternate material. Today, an equivalency assessment is required along with individual studies. Equivalency assessments and associated studies, by design, foster open discussion about process changes that may affect each party, and, at the same time, set the stage to properly implement changes that may be required to ensure a successful transition to new material.
Summary: How to prepare for raw material changes
To help prevent raw material changes from undermining your manufacturing operations, follow two basic tenets:
1. Work in partnership with your raw material suppliers. Let them know upfront that you need to know if they make changes to their products, as well as to fully understand the changes made. And;
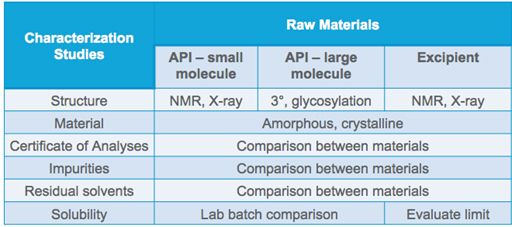
2. Do your due diligence before accepting any new material. Conduct equivalency tests using both existing and pending substances. Compare them side by side, from characterization studies to batch and bulk production of finished dosage forms.
Following this best-practice model will serve to maintain the integrity of your drug when raw materials change.