
Quality Risk Management 101: Overcoming Common Challenges In QRM Implementation
By Kelly Waldron, Ph.D., ValSource, Inc.

This article marks the last in a series of six intended to provide a holistic primer on the field of quality risk management (QRM). The first article, Quality Risk Management 101: Risks Associated with Medicinal Products, discussed the difference between intrinsic and extrinsic risks and clarified the scope of QRM efforts. It was followed by Quality Risk Management 101: A Brief History Of Risk Management In The Regulation of Medicinal Products. Quality Risk Management 101: ICH Q9 In Context” offered a critical discussion of the QRM process proposed by ICH Q9, while Quality Risk Management 101: QRM and the Product Life Cycle [link] discussed the relationship between QRM and quality by design, the pharmaceutical quality system, and post-approval change management. Quality Risk Management 101: A Review Of Required Reading For QRM Practitioners discussed some pivotal publications that all QRM practitioners should be familiar with. This article explores challenges with QRM implementation that are shared by many within industry.
After 10 years of quality risk management (QRM) experience in the pharmaceutical, biopharmaceutical, and medical device industries, I have had the opportunity to discuss QRM implementation with practitioners across the globe. Over time, I have identified several challenges that appear to permeate the industry as a whole, each of which poses a unique and very real challenge to the fully effective implementation of a QRM program. This article explores each of these challenges and explains the ways in which they might sabotage QRM efforts.
Misconceptions Regarding Quality Vs. Compliance, Or “Taking A Conservative Approach”
The first theme is confusion between compliance with ICH Q9 and effectiveness of QRM implementation. These, of course, are two very different concepts; compliance is often defined as “following regulation,” or applying the GMPs, while effectiveness in QRM stems from applying the regulations in a way that has tangible benefits such as product quality improvement, quality systems improvement, and realization of business objectives. Over the years, I have formed a habit of asking industry practitioners to sketch their understanding of the relationship between quality and compliance using a Venn diagram format. Venn diagrams are graphs of interlocking circles that are often used to demonstrate the relationship between categories, including the relative size or contribution of each category (as depicted by the size of a given circle) and the level of similarities and differences between categories (as depicted by the extent to which the circles overlap). A pattern in these Venn diagrams emerged early on, and has been reinforced many times at multiple industry conferences. The vast majority of industry practitioners draw a diagram similar to that shown in Figure 1.
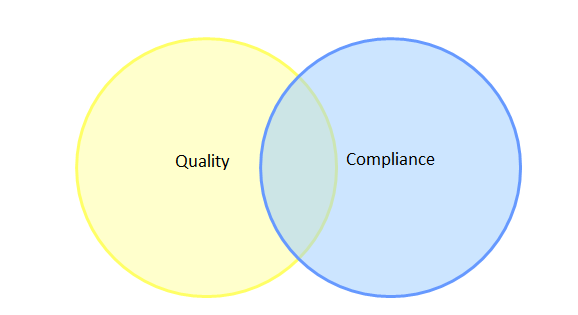
Figure 1: Typical Venn diagram sketched by industry practitioners to illustrate the relationship between quality and compliance
This diagram implies that most of industry believes there are aspects of quality that are unrelated to compliance, and more worryingly, that there are aspects of compliance that are unrelated to quality. With this being the paradigm under which some members of industry operate, it is not surprising that quality culture has become a topic of concern with regulators, since this opinion could embitter personnel to compliance and QRM-related activities if the value is not understood.
This poses a serious challenge to the enhancement of QRM effectiveness in industry, since the difference between quality and compliance (or compliance and effectiveness) is fundamental to understanding the role that QRM plays in the pharmaceutical and biopharmaceutical industries. ICH Q9 notes that “appropriate use of quality risk management can facilitate but does not obviate industry’s obligation to comply with regulatory requirements”1 — a tenet that (in my opinion) should be understood to mean that QRM is a mechanism through which compliance-related activities can be linked to product quality. In addition, QRM offers industry an opportunity to define what quality looks like for its patients, products, and businesses, beyond the basic requirements associated with regulatory compliance. As a result, the Venn diagram showing the relationship between quality and compliance through the lens of QRM looks more like that shown in Figure 2.

Figure 2: Venn diagram of quality and compliance through the lens of QRM
In this model, compliance is wholly encompassed by quality, such that all compliance-related activities likewise add to the quality of the product, and the circle representing quality has been enlarged based on the knowledge gained through QRM. This is the purpose of QRM; in ensuring that compliance supports quality and quality is based on risk management principles and practices, the patient is adequately supported.
A consequence of the misunderstanding of the role of quality risk management in protecting the patient has manifested with some members of industry claiming to use a “conservative approach” in lieu of QRM. Discussions with many industry practitioners reveal a misunderstanding that QRM need not be used in certain circumstances if a “conservative approach” is employed. One practitioner summed up the intent of this term with regard to validation, indicating that he did not apply QRM to determine what and how much to validate, because he validates “everything.” Seasoned QRM practitioners cringe at this statement, since it indicates a void of knowledge about the purpose of risk management. For example, validating “everything” circumvents any drive to distinguish between critical and noncritical elements, as identified in ICH Q8 and Q11, and therefore dilutes the amount of attention and resources spent assuring that elements critical to the patient are under control — an approach that is certainly not conservative with regard to the patient. It appears that some members of industry perceive QRM as a mechanism to do less, shrinking the amount of resources needed to perform an activity, rather than reallocating available resources to focus more on things that are critical and less on things that are not.
Insufficient Regulatory Guidance, Combined With Overly Prescriptive Regulatory Requirements
Many industry practitioners consider the lack of concrete, actionable guidance in ICH Q9 (and regional regulations adopted from this guideline) to be a challenge associated with QRM implementation. ICH Q9 outlines a framework for QRM and offers examples of how QRM can be applied but does not provide tactical information regarding how QRM can be used to fulfill these purposes. This challenge has been compounded by the eagerness of regulatory authorities to encourage industry to adopt QRM practices, publishing a flurry of requirements to use QRM to accomplish certain deliverables without sufficient guidance on how this should be accomplished within a QRM framework.
For example, a small group of delegates at a 2015 conference met after the day’s activities to discuss how their respective companies planned to implement the (then) recently released EU guideline “on the formalised risk assessment for ascertaining the appropriate good manufacturing practice for excipients for medicinal products for human use.” This document requires the use of a formal risk tool (hazard analysis and critical control point [HACCP] is suggested) to determine the rigor of GMP to be applied by suppliers of excipients and enforced by the drug manufacturer.2 The document lists 18 factors to be considered in the risk assessment, as follows:
- “Transmissible spongiform encephalopathy
- Potential for viral contamination
- Potential for microbiological or endotoxin/pyrogen contamination
- Potential, in general, for any impurity originating from the raw materials, e.g., aflatoxins or pesticides, or generated as part of the process and carried over, e.g., residual solvents and catalysts
- Sterility assurance for excipients claimed to be sterile
- Potential for any impurities carried over from other processes, in absence of dedicated equipment and/or facilities
- Environmental control and storage/transportation conditions including cold chain management, if appropriate
- Supply chain complexity
- Stability of excipient
- Packaging integrity evidence
- The pharmaceutical form and use of the medicinal product containing the excipient
- The function of the excipient in the formulation, e.g., lubricant in a tablet product or preservative material in a liquid formulation, etc.
- The proportion of the excipient in the medicinal product composition
- Daily patient intake of the excipient
- Any known quality defects/fraudulent adulterations, both globally and at a local company level related to the excipient
- Whether the excipient is a composite
- Known or potential impact on the critical quality attributes of the medicinal product
- Other factors as identified or known to be relevant to assuring patient safety”2
The group of delegates lamented the challenges posed by this guideline: the poor fit between many items on the list of required considerations and formal risk tools (including HACCP as the document had suggested), the number of individual risk assessments to be performed (one each per excipient per supplier), and the short timeframe for required implementation (roughly one year from the date of publication). Several delegates agreed that a tool such as risk ranking and filtering (RRF), also described in ICH Q9, would be a better fit than HACCP or FMEA (failure modes and effects analysis); other delegates pointed out that RRF is typically considered a less formal tool and would not meet the requirement that a “formalized” risk assessment be performed. One delegate expressed his wish that the guideline had simply included the expected format, so he could spend his time executing the approach rather than trying to define it. The informal meeting concluded with no harmonized agreement on the best path forward.
This anecdote is just one example of the struggles reported by QRM practitioners when trying to meet the detailed requirements of regional regulatory bodies within a more fluid, loosely defined QRM framework as offered by ICH Q9. The gap between an overly prescriptive “what” and an insufficiently prescriptive “how” has been identified as one of the obstacles preventing a more effective state of QRM implementation to be reached.
Excessive Numbers Of Risk Assessments
As suggested above, many industry practitioners cite the sheer number of risk assessments that have been created as a challenge in achieving a more mature state of QRM. Some note that regulators appear to expect a discrete risk assessment for every decision or GMP direction in which their companies proceed. Using the above example regarding excipients, a firm with five products, each having four excipients that can be purchased from a mere two qualified suppliers, would need to create and periodically review 40 risk assessments — just for the relatively narrow risk question regarding the level of GMP required of its excipient suppliers. Indeed, this trend can be seen in other areas as well; regulators expect risk assessments related to elemental impurities as described in ICH Q3D, Guideline for Elemental Impurities3, risk assessments related to viral or other contamination such as those implied (among other sources) in ICH Q5A(R1), Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin4 and FDA Guidance Sterile Drug Products Produced by Aseptic Processing – Good Manufacturing Practice5; risk assessments related to cross-contamination such as that suggested by EMA’s Guideline for setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities6; and so on.
These individual, narrowly construed risk assessments can quickly compound to the point of unmanageability. In December 2009, Wallace Torres at Roche told The Gold Sheet that in response to the 2007 public health crisis associated with chemical contamination of its popular HIV drug Viracept, “we performed more than 100,000 full FMEA analyses worldwide in the first year [following the initiation of the company’s QRM program].”7 While at the time this was a triumph of QRM implementation, I have since seen many examples where excessive numbers of risk assessments bogged down the QRM program and minimized value that can be extracted from the assessments, as time spent administering the program is time not spent gaining knowledge. It is my opinion that instead of using a “shotgun approach” to the creation of risk assessment, industry should focus on the creation of a strategic risk assessment library that minimizes the number of unique risk assessments performed while maximizing the scope and breadth of a given risk assessment to include multiple considerations of product quality, including those required by regulation.
Lack Of Resources To Focus On Risk Management
Many industry practitioners cite a lack of resources, including time and personnel, to focus on risk management as a potential obstacle in the way of further progress. Perhaps this concern is more appropriately characterized as a lack of managers’ willingness to deploy resources toward QRM, rather than a lack of availability of these resources. Formal risk management techniques such as FMEA often consume between 40 and 80 hours of work for a team of six to 12 people, not including the resources needed to prepare for the assessment or to track and implement risk control/mitigation actions. The challenge of resourcing is particularly acute at those companies with a “fire-fighting” culture, where personnel are largely (perhaps habitually) focused on solving existing problems rather than identifying and resolving potential risks. I suggest that management’s role in encouraging proactive behavior, including that required by effective QRM, is to identify and reward those individuals engaged in quality risk prevention, rather than exclusively rewarding those who solve “crises.”
Fear As An Obstacle To Implementing QRM
It is quite interesting that a primal emotion be listed as an obstacle preventing the successful implementation of QRM; however, this concept has indeed revealed itself. Many industry practitioners report a general reluctance within their organizations to embrace the transparency needed to perform QRM tasks, manifesting in several ways:
- Reluctance to analyze products, processes, and systems in a way intended to identify weakness, stemming from the fear that an urgent looming problem would be identified. Some practitioners liken this to a perception that “what we don’t know can’t hurt us,” pointing out that in most cases, QRM results in more work through the identification of mitigation activities.
- Uneasiness with the idea that, were weaknesses identified and documented, regulators would use the information to assign inspection observations. Some compare risk assessments with internal audit reports, which must be completed as part of a larger program but are generally not reviewed by inspectors so as to not discourage a firm from thoroughly identifying actual and potential problems, believing that risk assessments should be treated similarly.
- Discomfort with anticipated differences of opinion between the risk team that created a risk assessment and a third-party reader (whether internal or external to the company). Because QRM is often a subjective endeavor, it ought to be difficult to proclaim its outputs correct or incorrect without data to prove otherwise; however, some in industry indicate that their internal stakeholders often disagree with the analysis performed and conclusions drawn, while others express concern that an inspection observation might been received when an inspector believes that certain “rules” should have been applied to the scoring of individual risks where the risk team had felt otherwise.
Dr. Janet Woodcock, head of the CDER at the FDA, has also expressed concerns regarding a culture of fear, noting:
“Let me just step back another step and say — and this would also disturb some people — that I really think the culture of regulation that we had over the years, [produced] a kind of a fear relationship. And I am still told that industry is in a state of fear, many of them, of FDA. That kind of a fear relationship is not going to grow a quality culture, because there is a fear of adverse consequences… That is antithetical to the idea of a quality culture, where people own quality and say, ‘we can stand up to the FDA because we make a quality product and we know it and we monitor it and we are proud of it. That is our quality culture.’”8
The reluctance to embrace QRM based on these fears is indicative of a lack of risk maturity and a struggling company culture. Leadership within each company should strive to encourage such transparency and honest, open discussions regarding the identified vulnerabilities and the actions that may be taken to minimize them.
The challenges described in this article are industrywide problems and should therefore have industrywide solutions. I encourage those in industry who have successfully managed these challenges to share their experiences — speak at industry conferences, join QRM-related interest groups, and publish your experiences for others to learn from. Industry, academia, and regulators alike should strive to overcome these issues to enhance the effectiveness of QRM programs, and, most importantly, to deliver the highest quality to the patient, each and every time.
References:
- ICH. ICH Q9: Quality Risk Management. Jun 2005.
- EU. Guideline 2015/C 95/02. On the formalised risk assessment for ascertaining the appropriate good manufacturing practice for excipients for medicinal products for human use. March 19, 2015.
- ICH. ICH Q3D: Guidelines for Elemental Impurities. Dec 2014.
- ICH. ICH Q5A(R1): Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin. Sep 1999.
- FDA. Guideline for Industry: Sterile Drug Products Produced by Aseptic Processing – Good Manufacturing Practice. Sep 2004.
- EMA. Guideline for setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities. Nov 2014.
- Cox, B. Roche Builds Quality Risk Management Program in Response to Viracept Crisis. The Gold Sheet. Dec 1, 2009.
- International Pharmaceutical Quality (IPQ). Transcript of Dr. Janet Woodcock's speech on FDA's "Quality Revolution," given at the April 2015 ISPE quality metrics meeting. [Online] May 2015. [Cited: October 29, 2017.] https://www.ipqpubs.com/wp-content/uploads/2015/05/Woodcock-box-for-STOM.pdf.
About The Author:
 Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.
Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.