
A Framework For Quality Risk Management Of Facilities And Equipment
By Phil DeSantis

This two-part article focuses on risk management of facilities and equipment. It describes how a risk-based approach to facilities and equipment management fits into an integrated, effective quality systems structure. The principles discussed are equally applicable to all quality systems. Facilities and equipment represent a broad range of risk to product quality and are one of the key quality systems commonly identified in the pharmaceutical manufacturing industry.
The discussion of risk is limited here to quality: safety, purity, strength, and identity of product. Risk to health, safety, the environment (HSE), and business continuity are not considered; however, the principles discussed are fully applicable to those factors, while the risk controls applied may be different.
The first installment provided background and introduction to quality systems and quality risk management (QRM) and their relationship. In this second installment, facility and equipment risk are specifically addressed, and a framework for effective risk assessment and quality system application is proposed.
Facility And Equipment Risks
When equipment performs a function that changes the character of the material it contacts, it embodies risk to the quality of the final product. This function may be direct; for example, providing mechanical energy necessary to alter the physical or chemical nature of the material, i.e., a mixer. Indirect functions include those that do not contact the product directly but provide energy transfer or other functionality to change the conditions under which the process operates. Examples include jacket steam and cooling water, often grouped under the heading of “plant utilities.” It should be noted, however, that before an indirect function may be considered lower risk, it must be linked to a controlling and monitoring function that ensures that the desired critical parameter range is maintained. The instruments that perform the controlling and monitoring themselves have a direct effect on product quality and are considered to be of greater risk.
Systems or equipment that are used to support GMP operations but that do not embody at least one quality critical requirement (QCR) may generally be classified as low risk to quality without further assessment. Examples include jacket services, water pretreatment, and non-product contact compressed gasses.
Systems that include at least one QCR require further evaluation. The more detailed assessment must look beyond the system as a whole. By virtue of having at least one QCR, we can establish that a system has some direct quality impact and must be dealt with accordingly when applying quality systems. However, not every component in a direct-impact system is critical to quality. Therefore, risk assessment must look at each “major” component of a system to determine its risk. By “major,” we would define any component that is identified by a functional identification number (more commonly, a “tag” number) on a system schematic or piping and instrumentation diagram (P&ID). An example would be TIC-101 for temperature indicator controller #101. This would contrast to “parts,” which are not specifically identified on the P&ID but are built into the various components and identified on a separate parts list. Seals, shafts, and drive belts are examples of parts. (I might digress here to point out that a well-designed maintenance system will look as deeply at parts to assess risk of failure and design preventive maintenance programs. Such an assessment, however, focuses on overall performance and not just quality.)
Performing the risk assessment at the component (functional) level allows us to focus our quality systems on the parts of a system that actually influence QCRs and, therefore, affect quality.
Degrees Of Risk
The degree of risk for a system component may be determined by any number of well-known methods. Most, if not all, adhere to the International Conference on Harmonization (ICH) Q9 description of risk as:
Risk (R) = Severity (S) x Probability (P) / Detectability (D)
Some methods, notably failure mode and effects analysis (FMEA) and its derivatives, apply a numerical scale to each factor and then calculate an overall risk rating. Such methods can be very precise in determining apparent or projected risk. Other methods use an apparently more subjective approach, rating severity, probability, and detectability from low to high in fewer steps to determine overall risk.
Most firms I have worked with have chosen three or four tiers of risk (not including zero-risk equipment and systems that have no GMP functionality). We see Low/Moderate/High as well as Low/Low Moderate/High Moderate/High (terminology may vary).
Regarding the number of applicable quality risk categories, it makes no sense to have more categories than there are variations in quality systems workflows. More simply, the degree of risk should align with the risk control. We shall discuss this concept in more detail later.
It is valuable at this point to reemphasize our perception of quality. Remembering the first rule of quality risk management (QRM) and the focus on the patient, it is clear that risks associated with facility and equipment malfunction (or misapplication) are greater if they result in adulterated product (e.g., contaminated, sub-potent, mislabeled) reaching the patient. Risks associated with product rejection or lost batches are of lower degree because they are detected and dealt with before they reach the patient. Repeated failures of the latter kind are, however, an indication of poor quality systems and must also be corrected and action taken to prevent their recurrence.
The following matrix, derived from the Zurich Hazards Analysis (ZHA), is one I have used to assess facility and equipment risk at the component level. Red is high, yellow is moderate, and green is low risk. Different firms may assign differing risk levels to the various blocks, but this is representative of an acceptable approach.
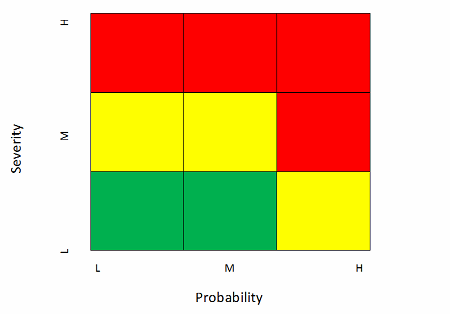
Note that I often discount detectability in my assessments. Most well-designed equipment and facilities will include monitoring and alarming devices that reliably detect any failure that can be readily measured. In addition, failures that manifest in product quality-critical attributes (CQAs) not meeting specification are often detectible in the laboratory. On the other hand, some of the higher severity risks, as we shall see below, are virtually undetectable. In summary, equipment and facility failures tend to be either detectable or not, so I choose to discount detectability and treat risks based on severity and probability, strongly weighted toward severity. If I had chosen a more equal weighting, the matrix might look like this.

In any case, I have found that facilities and equipment risk assessment is best performed by an experienced cross-functional team (operations, technical, quality) that will more than likely have predetermined risk even before the formal exercise using the chosen methodology. Therefore, the simpler the approach that meets the need, the better. The exercise is necessary to formalize and document the determination. Of course, the more of this is done prior to the application of other quality systems (e.g., change control, deviation management), the more effective the application of those systems.
The following matrix is an example of guidance that may be applied to risk assessment of facilities and equipment:

Application Of Quality Systems
The most important aspect of risk management is the application of appropriate controls once risks have been assessed. The proven approach to risk control is through quality systems. In the past, it has been a practice to apply quality systems uniformly across all levels of quality risk. This may have been understandable when good quality systems were in their infancy and variable workflows had not been developed. With GMPs for the 21st Century, however, the risk-based approach is essential.
Recalling QRM rule 2 (“If everything is critical, then nothing is critical.”), a uniform application of quality system workflow is wasteful and can be dangerous. Limited resources, especially among the quality unit, are rendered less effective by diverting their attention to issues that may have little relationship to product quality. For example, while attending to a change request on a low-risk system, an important high-risk situation may be overlooked. If you have not seen examples of this, you are indeed fortunate. My experience has been that application of risk-based workflows within quality systems has resulted in significant workload redirection among quality units responsible for change control and deviations, and among other important quality systems. It doesn’t mean they are less busy, but it does mean they are more focused on what counts.
Selecting quality systems most closely identified with facilities and equipment, I have developed a workable matrix that describes risks and suggests how the various quality systems may be applied. Notice that some risk levels follow the same quality system workflow. Of course, my suggestions are subject to debate; however, avoiding a dogmatic perspective (see QRM rule 3: “Dogma breeds ritual; ritual breeds waste.”) may allow firms to understand how risk management can be effectively applied within a structured quality systems program.
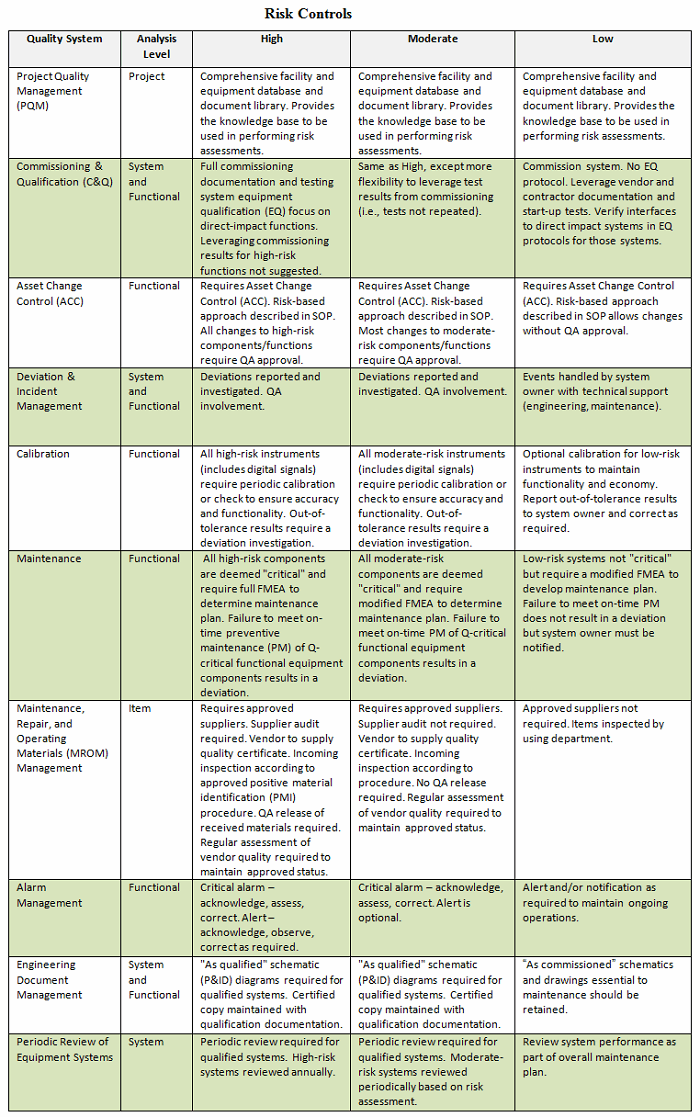
Conclusion
The “rules” of QRM introduced in this article may seem anecdotal, but they nonetheless represent reality. Keeping them in mind will make managing risks more efficient and more effective. Risk management in isolation may be valuable, but it can often be wasteful and misaligned with overall quality goals. By incorporating risk management into a structured quality system, the path to true product quality is ensured.
About The Author:
 Phil DeSantis is a pharmaceutical consultant, specializing in pharmaceutical engineering and compliance. He retired in 2011 as senior director of engineering compliance for Global Engineering Services at Merck (formerly Schering-Plough), where he served as global subject matter expert for facilities and equipment and on the Global Validation Review Board and Quality Systems Standards Committee. DeSantis is a chemical engineer, having received a BSChE from the University of Pennsylvania and an MSChE from New Jersey Institute of Technology. He has nearly 50 years of pharmaceutical industry experience. He is vice-chair of PDA Science Advisory Board and is active in ISPE. He has been a frequent lecturer and has published numerous articles and book chapters.
Phil DeSantis is a pharmaceutical consultant, specializing in pharmaceutical engineering and compliance. He retired in 2011 as senior director of engineering compliance for Global Engineering Services at Merck (formerly Schering-Plough), where he served as global subject matter expert for facilities and equipment and on the Global Validation Review Board and Quality Systems Standards Committee. DeSantis is a chemical engineer, having received a BSChE from the University of Pennsylvania and an MSChE from New Jersey Institute of Technology. He has nearly 50 years of pharmaceutical industry experience. He is vice-chair of PDA Science Advisory Board and is active in ISPE. He has been a frequent lecturer and has published numerous articles and book chapters.