
A Line-Of-Sight Approach To Continued Process Verification Planning
By Mike Long, Ph.D., ValSource, LLC

Utilizing language from the 2011 FDA process validation (PV) guidance, robust continued process verification (CPV) planning can guide an organization to:
- understand the sources of variation in the product and processes
- detect the presence and degree of variation
- understand the impact of variation on the process and ultimately on product attributes
- control the variation in a manner commensurate with the risk it represents to the process and product.
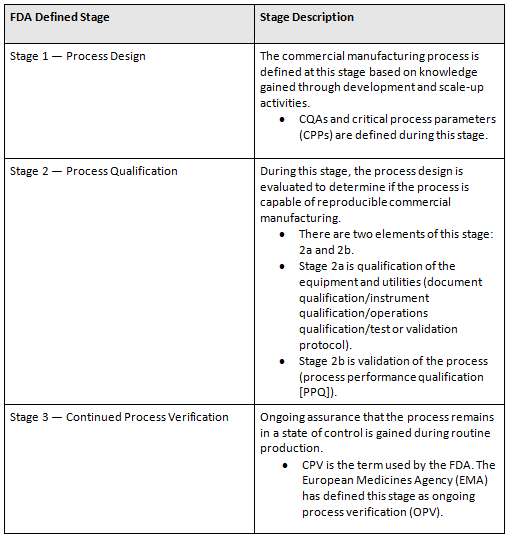
Any discussion on CPV planning needs to start at stage 1, process design. (See Table 1 for definitions.) It does not matter if the planning is for a new product, a tech transfer, or a legacy product. The components of stage 1 are critical. Identifying the relative severities of critical quality attributes (CQAs) and important material and process parameters is critical. Without them — for example, in the case of a legacy product — creation AND execution of CPV plans will not be possible.
Table 1: Process Validation Life Cycle Stages
CPV — Setting Up The Program
When setting up an organization’s CPV/OPV program, it is important to understand the types of products the firm manufactures, the scale of these manufacturing operations, and the number of batches manufactured per year. The approach used for an orphan product of which one batch a year is made will not be the same as a solid oral dosage product with 100 batches a year at multiple sites.
When setting up a CPV program, many organizations initiate their planning on products that have yet to be approved or introduced to market. However, the organization’s efforts should be initiated on products currently being manufactured and marketed. The overall risk (to the firm and to patients) is naturally greater for these products. If possible, the efforts should be in parallel. But for midsize to large companies with a large number of products, efforts should be placed on legacy first if prioritization is required.
CPV Requires Risk-Based Approach And Risk-Based Thinking
It is not uncommon for organizations to simply think of CPV as a new fancy term for statistical process control (SPC). While tools such as SPC are critical for the implementation of a CPV program, reliance on only those tools will not create a robust program and may even cause unwarranted reactions.
How is this? Remember, quality risk management (QRM) is embedded in the application of the process validation life cycle. It is noted throughout the 2011 FDA guidance as well as the EMA’s recent update of annex 15. The manner in which an organization plans, executes, and reacts requires an application of risk. And risk is defined as the combination of the probability of occurrence of harm and the severity of that harm:
Risk = Severity x Probability of Occurrence
SPC and other statistical tools used in a CPV program only provide one component of risk — probability of occurrence. — They do not provide the relative severity component of the risk calculation. If an organization does not have a robust QRM or risk-based thinking culture in place, overreaction to signals may occur. An organization’s CPV policies and procedures need to have QRM principles embedded in them so resources can be applied in a risk-based manner.
Thinking of using the ASTM E2500 standard for your process validation, but not sure where to start? Learn from Kelly Thomas's years of experience using this standard in the course:
Implementing the ASTM E2500 Validation Method: 10 Years of Key Learnings
Creating The Plan
As described in Table 1, there is short-term and long-term CPV. Each requires planning:
Initial planning components
- Description of the product and process with process map
- Scope of the CPV plan
- List the processes to be included.
- Is this a short-term or long-term plan?
- Is the plan for a new, tech transfer, or legacy (existing) product?
- List the batches used in the creation of the plan: at-scale clinical, PPQ, and production.
- Methods
- Include the risk-based statistical methods used to determine if enhanced sampling is required.
- Include the risk-based statistical methods used to determine reactions to signals (if these are not included in policies, procedures, or work instructions).
- General instructions
- Describe how samples will be taken and where data can be found (batch records, electronic laboratory information management systems, etc.).
- Describe any special storage conditions needed for samples.
- Describe in more detail which statistical tools may be used (e.g., graphical analysis, run charts, analysis of variance [ANOVA], variance components, SPC charting, and process capability analysis).
- Describe how many batches will be included in the plan.
- If enhanced sampling is required, describe how many batches will be required for these additional samples (n=5, n= 10, n=25?).
- Attributes
- List all critical attributes of the product and process, including:
- critical materials
- CQAs
- Provide relative severities for the CQAs.
- CPPs
- in-process controls (IPCs)
- Provide rationale for inclusion or exclusion of the attributes in the plan. For example:
- A CPP or IPC is a set point and does not have a variable output.
- The data output does not favor typical SPC charting and is covered under other monitoring programs (e.g., bioburden).
- There are not enough unique values to perform analysis (e.g., pH. For n=25 samples over N=25 batches, 10 of the values are 5.1 and 15 are values are 5.2.)
- Provide any additional analysis required for the justification of the plan (in appendices).
- List all critical attributes of the product and process, including:
An Important Note On Data Verification
The data used in the statistical analysis activities of the CPV PV program must be verified. While some organizations have the ability to electronically record lab results and transfer them into a statistical package to be analyzed, these organizations tend to be the exception. Most statistical analysis requires some manual entry into a statistical package whether it be entering it by hand or cutting and pasting. This manual movement of data and entry into stat packages has a risk of transcription error.
An organization needs to create processes and procedures for verification of the data used in the analysis. The approaches may differ company to company, but this is a critical step in assuring the continuous improvement decisions are not being made due to a transcription error. The person verifying the data used in the statistical analysis does not have to be a statistician. However, in conjunction with the data being verified as accurate, organizations should have a reviewer with statistical training engaged in the review and approval of a CPV report (and any other process validation life cycle document that has statistical analysis components to it).
Conclusions
Planning for CPV begins during development with the understanding of important product and process attributes to be monitored during the life span of the product. Robust processes and systems need to be implemented to execute the plan. Effective training on the life cycle, as well as statistical and risk-based thinking, is needed to realize the goal.
References:
- EudraLex, The Rules Governing Medicinal Products in the European Union, Volume 4, EU Guidelines for Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation, March 2015.
- U.S. Food and Drug Administration, Guidance for Industry – Process Validation: General Principles and Practices, January 2011.
- Long, “Risk and Statistics Serve as Tools for Solving Variation Riddles and Creating Robust Processes”, PDA Letter, Vol. XLIX, Vol. 3, 2013.
- Waldron, Greene, and Calnan, “Quality Risk Management: State of the Industry, Part 1. Has Industry Realized the Full Value of ICH Q9?”, Journal of Validation Technology, Vol. 20, No. 4, 2017.
- Baseman, Hardiman, Henkels, and Long, “A Line of Sight Approach for Assessing Aseptic Processing Risk: Part I”, PDA Letter, May 31, 2016.
- “QRM and Applications to the PV Lifecycle Approach” PDA Delaware Valley Chapter Meeting, September 30, 2014.
- Long, “Quality Metrics – What does it really mean?”, Contract Pharma, March 9, 2016.
- Long, “Driving Manufacturing Excellence”, PDA Education at Interphex, March 18, 2014, New York.
About The Author:
 Mike Long, Ph.D., is currently senior director of consulting services at ValSource, LLC. He is a past member of the Parenteral Drug Association’s (PDA’s) Science Advisory Board and co-leader of the PDA’s Quality Risk Management interest group. Long is the current lead of the PDA Applied Statistics interest group and co-leader of the PDA’s new CPV task force. Long is a frequent industry speaker/writer who has instructed courses in data analysis at Tufts University’s graduate engineering management program and taught risk management and quality systems in Regis College’s graduate program in regulatory and clinical research management. Long is a master black belt who earned a bachelor’s degree from Worcester Polytechnic Institute, a master’s degree from Tufts University, and a doctorate from Northeastern University.
Mike Long, Ph.D., is currently senior director of consulting services at ValSource, LLC. He is a past member of the Parenteral Drug Association’s (PDA’s) Science Advisory Board and co-leader of the PDA’s Quality Risk Management interest group. Long is the current lead of the PDA Applied Statistics interest group and co-leader of the PDA’s new CPV task force. Long is a frequent industry speaker/writer who has instructed courses in data analysis at Tufts University’s graduate engineering management program and taught risk management and quality systems in Regis College’s graduate program in regulatory and clinical research management. Long is a master black belt who earned a bachelor’s degree from Worcester Polytechnic Institute, a master’s degree from Tufts University, and a doctorate from Northeastern University.