
A Semi-Continuous Operations Model For Solid-Dose Manufacturing
By Ajay Babu Pazhayattil and Naheed Sayeed-Desta

The estimated timeline for a drug product from discovery to market is 10 to 15 years. However, less than 12 percent of drug candidates that enter clinical trials will ultimately receive FDA approval.1 The Orphan Drugs Act of 1983 ensured that there were adequate incentives offered to drug developers working on therapies to treat fewer than 200,000 patients in U.S.2 The initiative was very effective in correcting a segment that was largely neglected by research-based pharmaceutical corporations — today, there are more than 450 orphan drugs on the U.S. market. Orphan drugs were approximately 50 percent of the approved drug products based on FDA approval tracking. 3 As a result, the new small molecule generic pipeline will potentially comprise fewer high volume products.
This article will explore the evolving generic drug environment in the U.S. It will also describe an operations model for semi-continuous manufacturing of generic solid-dose products that will improve flexibility and enable just-in-time production.
The Current Generic Manufacturing Landscape
In August 2017, the FDA reauthorized the Generic Drug User Fee Amendments (GDUFA) to ensure patients have access to affordable and safe generic drugs.4 The FDA’s new commissioner, Scott Gottlieb, M.D., has devised strategies to minimize barriers to generic drug competition and bring down drug prices,5 and the impact has already been felt within the industry. As a result of GDUFA-related initiatives, the number of abbreviated new drug approvals (ANDAs) granted in 2017 is projected to be close to 756, almost 100 more than in 2016.6 The increased number of approved generic drug products and manufacturers in the market is expected to shift the demand curve and induce cost moderation. Accessibility of multiple generic sources will also drive down the volume blocks for competing drug manufacturers.
The recent acceleration in generic price erosion is partly due to a product approval surge for some smaller generic manufacturers.7 Many generic manufacturers maintain a diverse product portfolio to maintain a competitive edge in the market, as generic products make up the majority of the prescription market share (91 percent filled).8 FDA’s new risk-based policy and inspection concept of operations (ConOps)9 will have an impact on such generic pharmaceutical firms, as complex portfolios increase regulatory risks.
Innovator corporations continue developing strategies to maintain their niche market share. Developing new modified-release products, adding new IP protection to existing innovations, compensating for launch delays, etc. are some of the commonly used strategies making generic product development and launch difficult.10 Another disadvantage for generic manufacturers is the possibility of innovators developing newer drugs with similar mechanisms of action. The recent growth of approved biopharma products for multiple therapeutic categories is a threat to generic drug makers, as well. A prime example of these complications can be found in statins (atorvastatin, rosuvastatin, etc.), which are high-bulk generic drug products. The demand forecast for statins is somewhat uncertain, as it depends on market movements of Repatha (evolocumab), Amgen’s new monoclonal antibody for the treatment of high cholesterol.11
New pharmaceutical technologies such as continuous manufacturing, 3D printing, and combination products are also disruptors in the generics space. Also, personalized medicines and gene therapy business models are now being successfully developed — Kymriah, approved by the FDA in August 2017 for a rare form of leukemia, is a prime example.
The key driver to generic drug growth was the patent cliff. According to the QuintilesIMS Institute for Healthcare Informatics, the value of small molecules expected to lose patent exclusivity in developed markets between 2014 and 2018 is $121 billion. Post 2018, the highest revenue-generating blockbuster drug products will be large molecules.12
Further, generic drug manufacturers face mounting operational challenges. Existing high-volume batch manufacturing processes carry high maintenance costs, which affects a firm’s ability to invest in new or emerging technologies. Lack of clarity on potential industry-friendly regulations such as ICH Q8,13 which was intended to eliminate regulatory burden, has amplified the R&D expenses. Technology upgrades to ensure data integrity, for example, have added to product conversion costs. Limited expertise with advancements such as process analytical technology (PAT) has hindered implementation of efficiency improvement projects across the generic landscape.
Don't let your data integrity come into question. Attend this webinar to learn the current expectation for handling OOS investigations and assess your process for gaps- before the auditors do!
Laboratory Data Integrity: Current Expectations for OOS Result Investigations
Operations Management Strategy
A distinctive operations management strategy is needed to stay competitive amidst the radical changes occurring in the small molecule drug product manufacturing industry. Organizations must retain a large product portfolio to remain visible. They will also need to deliver products from their portfolio with a high supply chain velocity to meet customer expectations. This trend has become more prominent with the consolidation of wholesalers and pharmacy benefit managers in the U.S. market. Cardinal Health aligning with CVS Caremark,14 Walgreen Boots Alliance’s acquisition of nearly 2000 stores and three distribution centers from Rite Aid,15 and McKesson joining forces with Wal-Mart16 are some examples. Consequently, supply chain flexibility is a key element to success in the generic business.
Manufacturing operations are expected to increase throughput while simultaneously minimizing operational disruptions resulting from deviations or failures. Scaling products with variable batch sizes is an option; however, this incurs substantial R&D costs and increased CAPEX spending due to additional infrastructure and time requirements. Capacity optimization across global operations is typically conducted by larger establishments. The best solution would be manufacturing operations that provide ample supply chain flexibility.
Small molecule pharmaceutical manufacturing operations include individual processes such as dispensing, milling, blending, compaction, wet granulation, tablet compression, encapsulation, and coating. When closely analyzing each of these processes, they can be categorized as a batch process, semi-batch process, or semi-continuous process. According to the FDA, in batch operations, all materials are charged before the start of processing and discharged at the end of processing (e.g., bin blending). In a semi-batch process, the materials are added during processing and discharged at the end of processing (e.g., wet granulation). In a semi-continuous process, the material is simultaneously charged and discharged from the process, but for a discrete time period (e.g., tablet compression or roller compaction).17
In semi-continuous processes, the equipment set up impacts the entire run, and potential inherent process drifts should be understood and risk mitigated as part of development. Semi-continuous processes have the benefit of flexible batch sizes, as the run time is only affected by the equipment capability. Since the operational time is inconsequential, product quality is consistent and reliable from start to finish. In addition, semi-continuous operations such as roller compaction and tablet compression do not incur scaling costs. Therefore, we suggest that manufacturers should set a goal of converting batch and semi-batch processes to semi-continuous.
The switch to semi-continuous is now feasible, thanks to the availability of new processing technologies, such as the semi-continuous coater. Another example is the semi-continuous twin screw granulator, which requires feeders and establishing mean residence time distribution. Such an arrangement provides the flexibility to scale the batch size up or down based on market demand and to minimize the scale of characterization studies. Such technology modifications will impact the current infrastructure, though only nominally, because the new technology and recommended capacity require a smaller footprint than traditional batch technology.
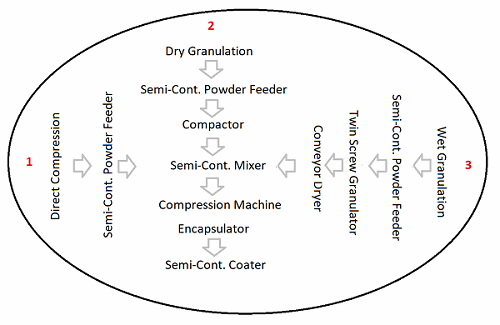
Unlike continuous manufacturing, where the entire process stream is held until completion of the campaign,18 the proposed semi-continuous process allows for flexibility in planning and scheduling the equipment for different processes, thus minimizing opportunity costs. The control system for the equipment is responsible for ensuring that the product is manufactured within the established process parameters. The at-line, on-line, or in-line PAT analyzer collects quality attribute data or chromatograms, which are converted to actionable information by the chemometric models. A locally available programmable logic controller (PLC) ensures the critical process parameter controls are conveyed to a central monitoring system, such as supervisory control and data acquisition (SCADA), which can also receive quality results from the PAT and multivariate statistical trending. A harmonized and integrated electronic control system ensures a high level of manufacturing control and awareness across the plant.
Implementing such complex systems would require a collective effort from the equipment provider, PAT instrumentation provider, software developers, and the drug formulator. As for future readiness, the same systems and lines can be converted to a fully continuous manufacturing process by establishing a mean residence time distribution, implementing a PAT feedback loop, and introducing advanced robotics to minimize the downtime between processes. Optimal product formulation strategies such as using multifunctional excipients should be considered. Advanced process modelling software can enable faster decision making, reduce R&D experimental costs, and allow for process design enhancements.
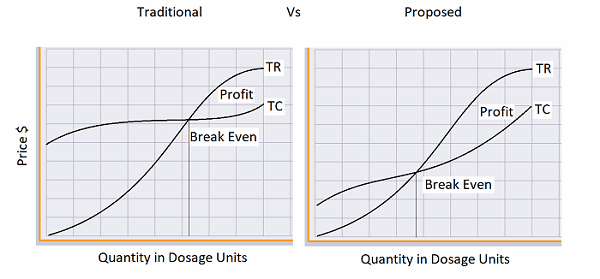
Assuming that the total revenue (TR) from the product is a constant curve, the current batch process methodology of installing multiscale equipment will increase the total costs (TC), thus taking longer to achieve break-even. With the proposed semi-continuous strategy, the initial total costs will be substantially minimized. Based on cost-volume-profit (CVP) analysis, the semi-continuous operating model will increase a firm’s profitability. Further, when there is a volume increase, the firm will be able to minimize marginal costs with the suggested model. The marginal cost slope will not be as steep, based on the fact that the infrastructure is already built-in, as well as the fact that added equipment will be smaller in scale, incurring minimal capital expenditure compared to the traditional batch processes.
Conclusion
The proposed manufacturing operations model converting existing batch processes to semi-continuous processes for solid dose manufacturing sites also involves application of integrated PAT, statistical process control (SPC), and predictive modeling software technologies. Implementing fully continuous manufacturing lines would result in engaging multiple fixed assets until the end of the manufacturing process, and hence may not be a desired option for manufacturers just yet. The semi-continuous strategy will improve production planning flexibility, enabling just-in-time manufacturing and injection of orders based on market demands, while avoiding backorder scenarios. Manufacturing of drug products only as required would allow for minimal overstock and prevent short expiry situations that would have otherwise resulted in further erosion of margins. The semi-continuous model enables manufacturers to maintain larger product portfolios with flexible supply chain prospects. The batch size flexibility, minimal footprint, and prospective decrease in development costs provides a distinct advantage. The improved ability for capacity optimization and enhanced production controls will ensure supply chain reliability, resulting in customer satisfaction.
Note: This article was prepared by the authors in their personal capacity. The opinions expressed are the authors’ own and do not reflect the view of their employer, government, or any agency with which they are affiliated.
References:
- PhRMA (2015). Biopharmaceutical Research & Development: The Process Behind New Medicines.
- U.S. Government Publishing Office. (2017). CFR Part 316, Title 21, Chapter 1, Subchapter D.
- Tribble, S.J., Lupkin, S. (2017). “Drugs For Rare Diseases Have Become Uncommonly Rich Monopolies.” NPR.
- U.S. FDA. Generic Drug User Fee Amendments (GDUFA). Retrieved Oct. 31, 2017 from https://www.fda.gov/ForIndustry/UserFees/GenericDrugUserFees/default.htm
- Gottlieb, S. (2017). “FDA Working to Lift Barriers to Generic Drug Competition.” FDA Voice.
- Pollock, B. (2017). Official August ANDA Approvals and Receipts Reported. Lachman Consultants. Westbury, NY.
- Altstedter, A., Hopkins, J.S., Modi, M. (2017). “With US Generic Drug Market in Chaos, Indian Upstarts Rise.” Bloomberg.
- PhRMA (2016). 2016 Biopharmaceutical Research Industry Profile.
- U.S. FDA Center for Drug Evaluation and Research, Office of Regulatory Affairs (2017). Integration of FDA Facility Evaluation and Inspection Program for Human Drugs: A Concept of Operations.
- Garde, D. (2017). “Pharma’s five favorite tricks to protect a monopoly.” STAT.
- U.S. FDA (2015). “FDA approves Repatha to treat certain patients with high cholesterol” (press release).
- Van Arnum, P. (2015). “On the Horizon: What to Watch for in 2015.” DCAT Value Chain Insights (VCI).
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (2009). ICH Harmonized Tripartite Guideline Q8 (R2): Pharmaceutical Development.
- Cardinal Health (2017). “CVS Caremark And Cardinal Health Announce Creation Of Largest Generic Sourcing Entity In U.S.” (press release).
- Walgreens Boots Alliance (2017). “Walgreens Boots Alliance Secures Regulatory Clearance for Purchase of Stores and Related Assets from Rite Aid” (press release).
- Walmart (2017). “McKesson and Walmart Announce Sourcing Agreement for Generic Pharmaceuticals” (press release).
- Chatterjee, S. (2012). FDA Perspective on Continuous Manufacturing, IFPAC Annual Meeting, Baltimore.
- Yu, L. (2016). Continuous Manufacturing has a Strong Impact on Drug Quality. FDA Voice.
About The Authors:
 Ajay Babu Pazhayattil has held key management roles with brand name, generic, and contract manufacturing organizations. He is an industrial pharmacist successful in delivering tangible results across multiple segments of pharmaceutical operations. Ajay has extensive experience in conducting global GMP audits and has been effective in conceiving, implementing, and promoting novel technology innovations based on sound scientific principles. His experience extends through solid-dose, liquids, and small and large volume parenteral dosage forms. Ajay is involved with industry organizations and have published multiple journal articles. Connect with him on LinkedIn.
Ajay Babu Pazhayattil has held key management roles with brand name, generic, and contract manufacturing organizations. He is an industrial pharmacist successful in delivering tangible results across multiple segments of pharmaceutical operations. Ajay has extensive experience in conducting global GMP audits and has been effective in conceiving, implementing, and promoting novel technology innovations based on sound scientific principles. His experience extends through solid-dose, liquids, and small and large volume parenteral dosage forms. Ajay is involved with industry organizations and have published multiple journal articles. Connect with him on LinkedIn.
 Naheed Sayeed-Desta has been responsible for providing strategic directions on life-cycle management for over 300 active solid-dose products. Naheed champions delivery of science- and risk-based approaches from traditional to novel manufacturing technologies. She is a proven leader in pharmaceutical manufacturing science and technology. Her expertise in providing pragmatic solutions for manufacturing operations is well recognized. She has been the lead author of journal articles. Naheed is an active contributing member of pharmaceutical industry organizations. Connect with her on LinkedIn.
Naheed Sayeed-Desta has been responsible for providing strategic directions on life-cycle management for over 300 active solid-dose products. Naheed champions delivery of science- and risk-based approaches from traditional to novel manufacturing technologies. She is a proven leader in pharmaceutical manufacturing science and technology. Her expertise in providing pragmatic solutions for manufacturing operations is well recognized. She has been the lead author of journal articles. Naheed is an active contributing member of pharmaceutical industry organizations. Connect with her on LinkedIn.