
A Straightforward, Risk-Based Approach to Better Quality Management System Design
By Mark F. Witcher, Ph.D., biopharma operations subject matter expert
As the complexity of biopharmaceutical products and their processes continues to increase, the quality management systems (QMSs) needed to support their development and manufacturing are becoming more difficult to design and implement. A QMS requires a wide variety of control strategies (CSs) to mitigate all risks to an acceptable level while keeping the manufacturing operation compliant with the wide variety of applicable regulations and guidelines (R&Gs). As used here, a QMS is a superset of a product quality system (PQS)1 because a QMS must also cover health, safety, and environmental (HSE) risks, requirements, and regulations. As an example, personnel safety for some products may be as important in CS development as patient safety and efficacy. This article proposes a fundamental change in the way QMSs are developed and implemented to build efficient, effective, and compliant QMSs in the 21st century.
systems (QMSs) needed to support their development and manufacturing are becoming more difficult to design and implement. A QMS requires a wide variety of control strategies (CSs) to mitigate all risks to an acceptable level while keeping the manufacturing operation compliant with the wide variety of applicable regulations and guidelines (R&Gs). As used here, a QMS is a superset of a product quality system (PQS)1 because a QMS must also cover health, safety, and environmental (HSE) risks, requirements, and regulations. As an example, personnel safety for some products may be as important in CS development as patient safety and efficacy. This article proposes a fundamental change in the way QMSs are developed and implemented to build efficient, effective, and compliant QMSs in the 21st century.
A QMS is a set of interconnected CSs that must meet the following three goals or design criteria:2
- Provide control – Establish and maintain control over the process or activity to minimize to an acceptable level all risks and assure the CS performs as required to meet all the process’ design criteria.
- Prove control – Provide documented evidence to a skeptical outside group that the control objectives were met.
- Improve control – Should control or proof of control not be met, provide mechanisms for quickly returning the process or system to a state of control and improve the CS to assure that control will be maintained in the future.
The requirement for always simultaneously achieving all three CS goals separates biopharmaceutical manufacturing from most other manufacturing operations. An integrated QMS must be designed to simultaneously achieve all three goals, especially the third goal. The goal of improving control requires that both root cause analysis (RCA) control strategies and corrective and preventive action (CAPA) be integrated into all other CSs. RCA and CAPA for continuous improvement require identifying and correcting both near misses as well as deviations from control objectives or realized risk consequences.
The prevailing or conventional methods for developing QMSs have evolved over many decades, beginning with very simple chemical products that were relatively easy to regulate and control. Because the products and their intermediates could be tested for manufacturing deficiencies, the initial CS approach consisted mostly of meeting the first two goals of providing control by documented testing of intermediates and the product. As products and their processes have become more complicated, it has become increasingly more difficult to state the required manufacturing practices as straightforward adequate and sufficient standards.3
Conventional QMS Design Approach
The usual approach is based on achieving compliance by reviewing and understanding the regulations. Regulations are by necessity fairly vague and subject to various interpretations, as they are applied to a wide variety of different situations, products, and processes. Regulations are primarily aimed at setting minimal acceptable benchmarks based on sufficient and adequate criterion and are not intended as a design standard. Defining minimal acceptable standards for complex biologics is very difficult. In some cases, due to high uncertainty and unknown factors, it may be appropriate to go well beyond what might be considered as a minimal acceptable standard. As the need for ever more comprehensive standards has increased, the difficulty of interpreting the regulations has also increased. To aid the industry in using regulations as a design criterion for QMS development, regulatory agencies began formulating guidelines. However, because the agencies do not want to be overly prescriptive, which could result in excessive complexity and possible misapplication to specific circumstances, the guidelines, too, are also intended to be flexible for a wide variety of settings and situations.
When compounded with the ever-widening types of products and therapeutics, from large biological molecules to advanced therapeutic medicinal products (ATMPs), the number and nature of the R&Gs has exploded into a large collection of interacting, overlapping, and occasionally inconsistent documents that are nearly impossible to completely and comprehensively read, review, and understand. As an example, good tissue practices (GTPs)3 reads very much like GMPs,4 with nearly identical concepts, approaches, and requirements. If handling and processing the tissue is viewed as manufacturing a product, then GTPs and GMPs have virtually the same objectives because they control very similar risks.
The current approach of using the myriad of R&Gs for QMS development is summarized in Figure 1. Given the complex products and processes, understanding all the R&Gs that might apply to a specific process and product over its development and manufacturing life cycle can be very challenging.
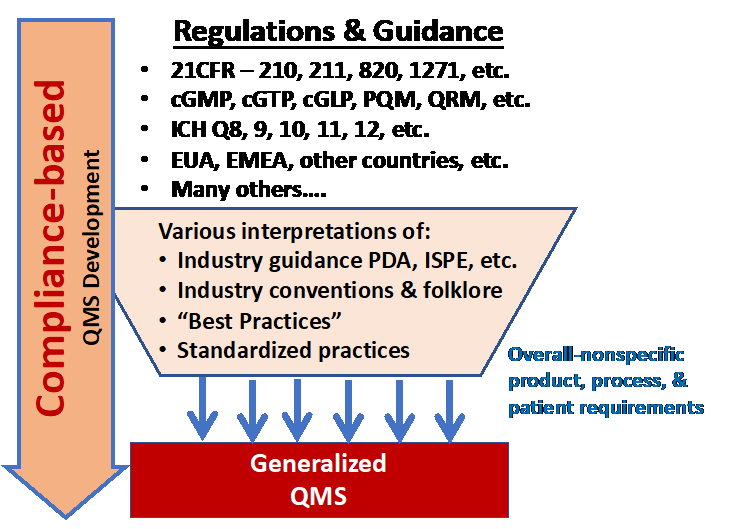
Figure 1: The conventional method of developing a QMS is to derive the necessary control strategies from government regulations and guidelines (R&Gs). The many R&Gs are processed through a wide variety of interpretations and viewpoints for assembly into a QMS designed to control development and manufacturing. The resulting QMS is typically a generalized system designed to control a wide variety of situations and risks.
To compound the problems, many people do not identify, read, and study all the applicable R&Gs. Thus, many QMSs are based on an incomplete set of regulatory standards, and, as a result, many QMS designs are based on what companies have seen in the past for other products and processes. Many companies have also attempted to standardize their QMSs to achieve uniformity across many different products, processes, and manufacturing facilities.
Page 3 of the FDA’s 2006 Guidance for Industry: Quality Systems Approach to Pharmaceutical CGMP Regulation includes the following statement:
A quality system adopted by a manufacturer can be tailored to fit the specific environment, taking into account factors such as scope of operations, complexity of processes, and appropriate use of finite resources. 5
Of course, the central issue is how to tailor the QMS to achieve appropriate use of finite resources based on the scope of operations and complexity of the processes.
If the primary design approach for building QMSs by using the very large number of complex R&Gs, as shown in Figure 1, is to change to an approach designed to control only the specific product, process, and facility risks, can we build more efficient and effective QMSs?
The Risk-Based QMS Design Approach
The alternative approach is to build the QMS based on identifying and evaluating the specific risks the product, manufacturing process, employees, and patients will be exposed to during development and manufacturing. The risks can be identified using the basic tools shown in Figure 2 and briefly described below.
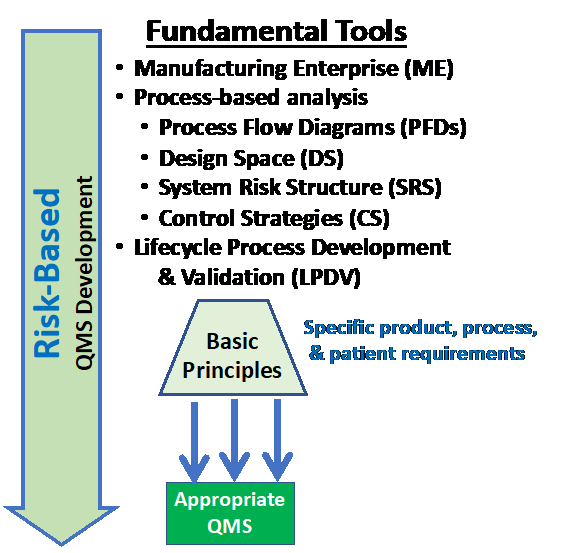
Figure 2: An appropriate QMS can be developed using basic tools and paradigms that focus on using a risk-based approach to define the necessary control strategies as part of the manufacturing enterprise (ME).
The QMS is a composite of a large number of control strategies or control systems that work together to control all the ME’s risks. Four tools or approaches can be used to identify and manage all the risks to the ME and the product that might reasonably occur and then build the necessary CSs to control those risks.
4 Fundamental Tools
The four basic tools start with understanding how an ME is organized. By identifying all the processes needed to manufacture a product using a process-based analysis, the risks associated with all the processes can be identified, evaluated, analyzed, and, when necessary, mitigated using system risk structures (SRSs).6 After the risks have been analyzed, the unacceptable risks can be controlled or mitigated by developing or improving CS processes using the life cycle process development and validation (LPDV) tools to control the risks to an acceptable level.2,7
1. Manufacturing Enterprise
All product manufacturing, from process development through commercial manufacturing, comes from a manufacturing enterprise composed of three elements: manufacturing process, facility, and infrastructure.2
- Manufacturing Process – The unit operations (UOs), equipment, instruments, piping, and materials used to manage the product. A quick definition might be all the systems that directly handle or influence the production of the product.
- Facility – The room layouts, including the flow of materials, personnel, equipment, etc. and the features surrounding the manufacturing process. Also included are the central or common utility systems that supply raw materials, such as WFI.
- Infrastructure (QMS) – The operating and manufacturing procedures, practices, policies, and other support systems that control the operation of the manufacturing process and facility elements.
To summarize, the ME can be described as a manufacturing process contained and supported by a facility under the control of the infrastructure.
2. Process-based analysis
All three ME elements can be subdivided into processes. These processes form a network of interdependent systems that control every aspect of making the product. These processes can be organized using process flow diagrams (PFDs) to map the connections and how the processes relate and interact with each other. For the manufacturing process described as a network of unit operations, a design space (DS) for each UO can be developed that places all the process inputs into either operating parameter, input material attribute, or equipment parameter categories that identify the threats to the UO’s outputs described as quality (product) attributes and process performance responses (output parameters).2
3. System Risk Structures (SRS)
The PFDs can then be used as the foundation for building the required SRSs to describe how input threats to a process might cause various risk consequences.6 The SRS can then be analyzed by a team of experts to determine the likelihood of a risk consequence’s occurrence using prospective causal risk modeling (PCRM).7 The subject matter experts analyze the likelihood of the threats happening as an input to the process, combined with the likelihood of the threat passing through the process because the threat cannot be adequately controlled, thus resulting in the occurrence of a realized risk. Risks that are unacceptably likely to occur are then controlled or mitigated by changing or adding CS processes to prevent the threat from occurring or preventing the threat from resulting in the risk’s occurrence.
4. Life Cycle Process Development & Validation (LPDV)
All processes, whether they are part of the ME’s manufacturing process, facility, or infrastructure element, have four basic questions:2
- What is the goal of the process’ design? – Define (Stage #0)
- How will the process achieve the goal? – Design (#1)
- Will the process design work? – Qualify (#2)
- Did the process’ execution work? – Operate & Verify (#3)
The four questions are common to every process, from SOPs, unit operations, equipment, cleanroom operation, personnel training, material flows through a layout, etc. The most important stage is the design stage (#1), where the following two execution stage questions are answered using Quality by Design (QbD), resulting in a nearly fully developed and validated process from the design stage forward. The four-stage LPDV paradigm is an evolution of the FDA’s groundbreaking process validation guidance.8
The four tools described above provide a relatively straightforward sequential paradigm for identifying, dealing with, and documenting a wide variety of risks as the basis of building the necessary CSs to control and accept the various risks.
The All-Important Final Step
All QMS elements must be compliant with all regulations. The final step is to review the risk-based QMS against the various R&Gs to make sure they meet the regulatory requirements and are consistent with appropriate guidelines. Although the regulations and guidelines for all the various QMS elements are frequently redundant, vague, and somewhat confusing, they still contain a lot of wisdom and important considerations for building a comprehensive and effective QMS. A thorough knowledge of the appropriate regulatory documents is essential and can be useful supporting information. If the QMS has been designed using the fundamental tools and basic risk mitigation and control concepts, checking its elements against the regulatory documents becomes much easier. Any gaps can be identified and resolved by going back to the SRSs and adding or modifying the CS processes appropriately to provide the necessary control and compliance.
Conclusion
Given how entrenched the current QMS development approaches shown in Figure 1 are, switching to a more basic approach shown in Figure 2 will be difficult for many. The recommended approach is to start small and build systems throughout the design process as process flow diagrams , design spaces, and system risk structures are constructed and reviewed. During the design, the interactions between the ME elements will require additional CSs be developed and enhanced to complete the necessary control of interacting risks. The new method is unlikely to initially save time and effort, but the approach is recommended because it will produce better QMSs and eventually save time and effort over the long haul.
However, the primary benefit will come from the documentation left behind from using the methods. SRSs, when combined with appropriate risk registers and summaries, provide effective documentation of the QRM’s design for internal company review and consensus as well as regulatory agencies. SRSs can be used to effectively describe the flow of identified threats through CSs to show remediation of risk consequences. In addition, the use of LPDV provides additional documentation, especially when QbD exercises are used during Stage #1 design activities, of how the CS was developed and implemented by modifying or adding control processes to the PDFs and SRSs. One of the primary problems with compliance based QMS designs is that there is virtually no specific documentation as to why or how things are designed to control the various risks other than citing the various R&Gs.
References:
- FDA Guidance for Industry – Q10 Pharmaceutical Quality Systems, April 2009.
- Witcher MF. Integrating development tools into the process validation lifecycle to achieve six sigma pharmaceutical quality. BioProcess J, 2018; 17, https://doi.org/10.12665/J17OA.Witcher.0416
- FDA Guidance for Industry – Current Good Tissue Practice (CGTP) and Additional Requirements for Manufacturers of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps), Dec. 2011.
- FDA Guidance for Industry – Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients Guidance for Industry, Sept. 2016; and others
- FDA Guidance for Industry – Quality Systems Approach to Pharmaceutical CGMP Regulations, Sept. 2006.
- Witcher MF. Analyzing and managing biopharmaceutical risks by building a system risk structure (SRS) for modeling the flow of threats through a network of manufacturing processes. BioProcess J, 2017; 16. https://doi.org/10.12665/J16OA.Witcher
- Witcher MF. Estimating the uncertainty of structured pharmaceutical development and manufacturing process execution risks using a prospective causal risk model (PCRM). BioProcess J, 2019; 18. https://doi.org/10.12665/J18OA.Witcher
- FDA Guidance for Industry – Process Validation: General Principles and Practices, Jan. 2011.
About The Author:
 Mark F. Witcher, Ph.D., has over 35 years of experience in biopharmaceuticals. He currently consults with a few select companies. Previously, he worked for several engineering companies on feasibility and conceptual design studies for advanced biopharmaceutical manufacturing facilities. Witcher was an independent consultant in the biopharmaceutical industry for 15 years on operational issues related to: product and process development, strategic business development, clinical and commercial manufacturing, tech transfer, and facility design. He also taught courses on process validation for ISPE. He was previously the SVP of manufacturing operations for Covance Biotechnology Services, where he was responsible for the design, construction, start-up, and operation of their $50-million contract manufacturing facility. Prior to joining Covance, Witcher was VP of manufacturing at Amgen. You can reach him at witchermf@aol.com or on LinkedIn.
Mark F. Witcher, Ph.D., has over 35 years of experience in biopharmaceuticals. He currently consults with a few select companies. Previously, he worked for several engineering companies on feasibility and conceptual design studies for advanced biopharmaceutical manufacturing facilities. Witcher was an independent consultant in the biopharmaceutical industry for 15 years on operational issues related to: product and process development, strategic business development, clinical and commercial manufacturing, tech transfer, and facility design. He also taught courses on process validation for ISPE. He was previously the SVP of manufacturing operations for Covance Biotechnology Services, where he was responsible for the design, construction, start-up, and operation of their $50-million contract manufacturing facility. Prior to joining Covance, Witcher was VP of manufacturing at Amgen. You can reach him at witchermf@aol.com or on LinkedIn.