
Advanced Process Risk Assessment And Control Strategy Development In Solid Oral Dosage Forms
By Dileep Boinipally, director of drug product formulation, Metsera

Effective risk assessment and control strategies are essential to the successful development of pharmaceutical solid dosage forms. As regulatory bodies increasingly emphasize quality by design (QbD), manufacturers must leverage scientific tools to identify and mitigate risks impacting critical quality attributes (CQAs) of the drug product. This article synthesizes the scientific development and manufacturing process risk assessments for four anonymized model drugs that eventually received FDA approval. Each model represents a distinct manufacturing strategy (granulation, direct compression, or capsule filling) and showcases how structured experimentation — particularly design of experiments (DoE) — combined with formulation optimization and process understanding mitigates high-risk variables and enables consistent product quality. The focus is on their process risk assessments, control strategy development, and how QbD principles1,2 were applied to transform manufacturing challenges into reproducible, validated processes.3
ICH guidelines Q8 through Q11 emphasize a science- and risk-based approach to pharmaceutical development, where process understanding is coupled with appropriate control strategies.4,5 Key CQAs such as content uniformity, assay, dissolution, and degradation products are systematically addressed through the development of control strategies rooted in pharmaceutical science.
Methodological Framework
Each model product underwent a multistage risk assessment framework, including:
- initial risk identification via FMEA5,6
- formulation and process optimization using DoE (full or fractional factorial)1,2
- verification through scale-up and registration batches3
- final risk reassessment post-data integration5,6
Process parameters were linked to CQAs through mechanistic and empirical understanding. Experiments targeted design space definition, allowing formulation and manufacturing to be executed within preestablished safe operational ranges.
Scientific Case Studies
Model Drug 1: Multi-particulate Extended-Release Capsule
Formulation Strategy: Model Drug 1 was designed as an extended-release dosage form using compressed tablets filled into capsules. The granules were prepared via top-spray fluid bed granulation, followed by drying, compression, and encapsulation.
Initial Risks:
- High risk to dissolution due to sensitivity of granulation parameters and tablet surface area.
- Medium risk to content uniformity during encapsulation due to tablet breakage and fill weight variability.
Scientific Rationale and Process Design:
- Granulation variables (product temperature, spray rate, atomization pressure) were optimized via 23⁻¹ fractional factorial DoE.1
- Acceptable dissolution ranges were identified (e.g., 5%–25% at 1 hour, 40%–70% at 4 hours, not less than 75% at 12 hours).
- A full factorial DoE was further conducted on binder and lubricant levels to evaluate their influence on erosion rate and dissolution profile.1
Control Strategy Highlights:
- Tablet thickness was controlled to maintain consistent surface area.
- An encapsulator with 100% fill weight verification and visual inspection to minimize breakage was used.
Final Outcome: Integration of process controls and material specifications significantly reduced risks to CQAs, allowing reproducible performance even at scale-up levels.
Model Drug 2: Immediate-Release Capsule via Direct Blend
Formulation Strategy: This product employed a direct blending and encapsulation process, with the API representing only 2.67% w/w of the blend — a known challenge for uniformity.
Initial Risks:
- High risk to content uniformity and assay due to poor flow and low drug load.7
- Medium risk to dissolution from lubricant distribution.
Scientific Rationale and Optimization:
- One factor at a time (OFAT) design followed by full factorial DoE,1 assessed interaction between excipients, including pregelatinized starch, sodium lauryl sulfate, and magnesium stearate.
- Targeted particle size (D90 < 10 µm) improved adsorption of API onto excipient surfaces.
Key Scientific Observations:
- Pregelatinized starch promoted ordered mixing, reducing standard deviation of blend assay to <3.0%.
- Sodium lauryl sulfate (0.9%–1.1% w/w) was critical for fast wetting and dissolution.
Control Strategy Highlights:
- Tight specifications were established for excipient grades.
- In-process weight checks post-encapsulation minimized variability to <1.5%.
Final Outcome: Process optimization enabled consistent blend and capsule performance. Residual risk to dissolution was controlled through surfactant-lubricant balancing.
Model Drug 3: Immediate-Release Tablet via Direct Compression
Formulation Strategy: A tablet based on direct compression was pursued to simplify manufacturing and reduce processing time.
Initial Risks:
- Medium to high risk to content uniformity due to direct compression of a high-dose drug.7
- Medium risk to dissolution due to lubricant overmixing.
Scientific Strategy:
- API particle size and flow properties were matched with filler silicified microcrystalline cellulose (SMCC) to ensure compressibility.
- A DoE matrix was used to optimize magnesium stearate and binder concentrations and identify interactions affecting dissolution.
Key Technical Findings:
- Over-lubrication delayed tablet erosion, compromising dissolution.
- Target blending time was established using OFAT screening,5 then confirmed in pilot batches.
Control Strategy:
- Real-time compression monitoring of tablet weight, hardness, and thickness was conducted.
- NIR-based in-line monitoring was considered for blending uniformity.
Final Outcome: All final batches consistently met USP criteria (assay, CU, dissolution) and were validated for scale-up.
Model Drug 4: Dose-Proportional IR Tablets (Multiple Strengths)
Formulation Strategy: Model Drug 4 was developed as a dose-proportional series (10 mg to 150 mg) using a common blend strategy, leveraging previous validation data from an approved 25 mg strength.
Initial Risks:
- Medium risk to uniformity of dosage unit and assay due to varying fill volumes and compression profiles.7
- Low risk to degradation products and dissolution, as the formulation remained unchanged.
Scientific Control Development:
- Blend uniformity and particle size were ensured via validated screening (U.S. #40 sieve).
- Compression process optimization was conducted across pilot and production scale rotary presses, each assessed for turret speed and feeder variability.
Scientific Controls:
- Powder flow studies confirmed consistent blend density and compressibility.
- Dissolution and assay uniformity were verified through stratified sampling at all strengths.
Final Outcome: Final CQAs were met across strengths with <3.5% relative standard deviation (RSD) for assay and <1% weight variation. Process design demonstrated effective scalability.
Unified Learnings And Scientific Discussion
Role of Particle Size and Excipient Selection
- Model Drug 2 showed that low drug load requires micronizing or controlled particle size distribution (PSD) for optimal blend homogeneity.
- In Model Drug 3, PSD indirectly influenced dissolution by modulating surface area available for erosion.
Process-Formulation Interactions
- For Model Drugs 1 and 3, binder-lubricant interactions significantly affected dissolution. High binder delayed release and excessive lubricant slowed erosion.
- Model Drug 2 revealed that hydrophilic surfactants compensated for poor drug wettability.
DoE Application and Design Space1
- Use of full and fractional factorial designs enabled efficient multivariate experimentation, providing insight into synergistic or antagonistic effects.
- These studies supported design space justification, where variations within tested ranges did not adversely affect CQAs.
Final Risk Mitigation Strategies6
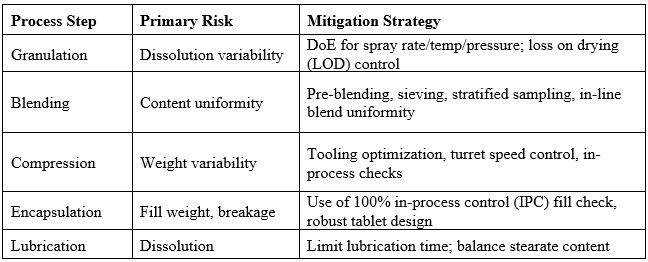
Conclusion
The integration of risk-based thinking, scientific experimentation, and robust control strategies enables pharmaceutical developers to mitigate process variability and ensure consistent performance in solid oral dosage forms. Across the four model drug products studied, effective application of DoE, blend and process characterization, and scale-up verification proved essential in translating development data into validated commercial manufacturing practices.
With global regulatory authorities emphasizing life cycle management and continuous process verification, the scientific rigor applied in these case studies sets a model for future product development and commercialization.
References
- LeBlanc D. Design of Experiments and QbD in Oral Dosage Forms. J Pharm Sci. 2021;110(3):1047–1060
- Rathore AS, Winkle H. Quality by Design for Biopharmaceuticals. Nat Biotechnology. 2009;27(1):26–34
- U.S. FDA. Process Validation: General Principles and Practices. Jan 2011
- ICH Q8(R2). Pharmaceutical Development. International Council for Harmonization, 2009
- ICH Q9. Quality Risk Management. ICH Harmonized Tripartite Guideline, 2005
- Nasr M. Risk-Based Approaches to Pharmaceutical Manufacturing. FDA, 2006
- USP <905>. Uniformity of Dosage Units. United States Pharmacopeia
About The Author:
 Dileep Boinipally is a seasoned pharmaceutical scientist and drug product development leader with extensive experience in formulation and manufacturing process development. He currently serves as director of drug product formulation at Metsera, Inc., leading the development of oral drug formulations. Previously, he was associate director of drug product development at EQRx International, overseeing the development of small molecule drug product development from early-phase clinical trials to commercialization. He has also held scientific leadership roles at Prothera Inc., Upsher-Smith Laboratories, and West-Ward Pharmaceuticals. Boinipally holds an M.S. in pharmaceutical engineering. Over the last 14 years, he has played a pivotal role in advancing drug candidates into clinics and toward commercialization.
Dileep Boinipally is a seasoned pharmaceutical scientist and drug product development leader with extensive experience in formulation and manufacturing process development. He currently serves as director of drug product formulation at Metsera, Inc., leading the development of oral drug formulations. Previously, he was associate director of drug product development at EQRx International, overseeing the development of small molecule drug product development from early-phase clinical trials to commercialization. He has also held scientific leadership roles at Prothera Inc., Upsher-Smith Laboratories, and West-Ward Pharmaceuticals. Boinipally holds an M.S. in pharmaceutical engineering. Over the last 14 years, he has played a pivotal role in advancing drug candidates into clinics and toward commercialization.