
An Analysis Of FDA FY2016 Drug GMP Warning Letters
By Barbara Unger, Unger Consulting Inc.

This article presents a detailed summary of the drug GMP warning letters issued by FDA in FY2016, as well as a comparison of trends in this area since FY2013. Firms should monitor publicly available health authority enforcement actions and trends as part of a comprehensive GMP intelligence program, because FDA expects firms to monitor these actions and to correct similar deficiencies at their site(s).

The data for FY2016, ending September 31, 2016, is based on drug GMP warning letters posted by the FDA no later than November 1, 2016. The appendix provides high-level details on each FY2016 drug GMP warning letter, identifying the product type covered in the letter, the issuing office, the number of deficiencies identified, the country in which the sites in question are located, and link to the complete letter.
The term “compounding pharmacy” also includes outsourcing facilities. Compounding pharmacies / outsourcing facilities are identified as a separate category based on their legal foundation. Although they are all located in the U.S., they are not combined with enforcement actions against U.S.-based pharmaceutical facilities. Outsourcing facilities were identified by FDA as drug manufacturing entities in November of 2013, and compounding pharmacies are governed by individual state laws.
Following are brief summaries of noteworthy findings and trends, followed by more detailed analysis in the subsequent sections:
- The number of drug GMP warning letters issued more than doubled over the previous year, from 42 in FY2015 to 102 in FY2016. (See Table 1 and Figure 1.)
- The compounding pharmacy/outsourcing facility segment continues to attract disproportionate enforcement attention from FDA, receiving more than 50% of the warning letters for the third fiscal year in a row. This suggests that firms are not learning from the enforcement actions taken against others. (See Table 1 and Figure 1.)
- The number of warning letters issued to API manufacturers and dosage manufacturers is approximately equal in FY2016, with a dramatic increase in the number of warning letters issued to API manufacturers over those issued in FY2015. (See Table 1 and Figure 2.)
- Excluding the compounding pharmacies and outsourcing facilities, FDA continues to focus enforcement actions outside the U.S. (OUS), where most generic drugs are produced. Over three times as many warning letters were issued to OUS firms compared to domestic firms. Firms in India and China received 71% of the warning letters issued to firms outside the U.S. Warning letters issued to sites in China increased from two in FY2015 to 15 in FY2016. (See Tables 1 and 2, and Figures 1 and 3.)
- The percent of warning letters that cite deficiencies in data integrity remains consistent at approximately 80% for OUS firms and reaches the same percentage for warning letters issued to sites in the U.S. in FY2016. This year saw a significant increase for U.S. warning letters citing data integrity deficiencies. (See Table 4 and Figure 4.)
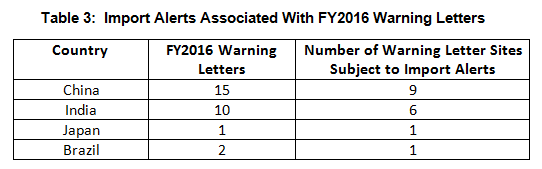
- Import alerts were associated with 17 of the 35 warning letters issued to OUS sites in FY2016. Firms in China and India that received warning letters were the subject of 15 of the 17 import alerts associated with warning letters. (See Table 3.)
- The interval between inspection and issuance of warning letters has increased over the past four fiscal years. When import alerts were put in place, they generally occurred in half the time required to issue the associated warning letter. (See Table 5 and Figure 5.)
Warning Letter Data
Table 1 shows that drug GMP warning letters more than doubled between FY2015 and FY2016. It also shows that the continued extraordinary focus on compounding pharmacies/outsourcing facilities continues, with more than 50% of warning letters issued to this group. FDA’s legal authority over these entities was clarified in the Drug Quality Security Act of 2013 (DQSA) and explains the explosion of enforcement action in this area beginning in FY2014.
Warning letters issued to API sites also have increased since FY2013 and have more than doubled from FY2015 to FY 2016. Similar increases occurred for sites that manufacture drug product.
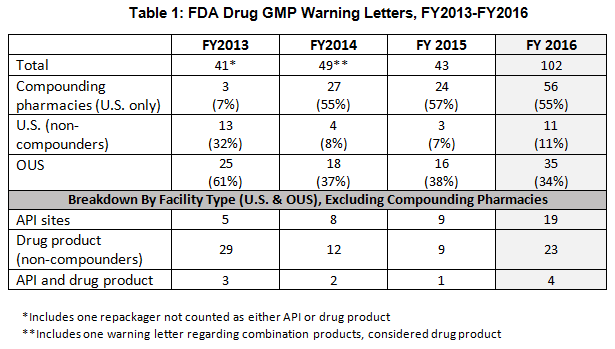
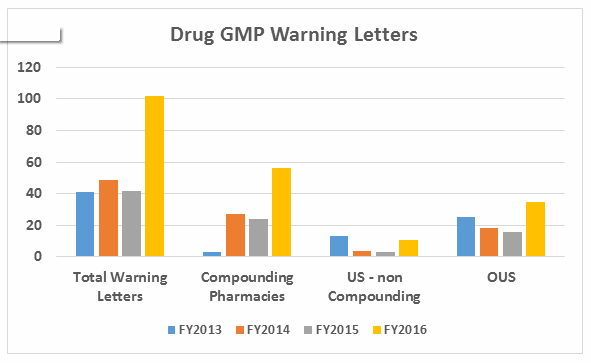
Figure 1
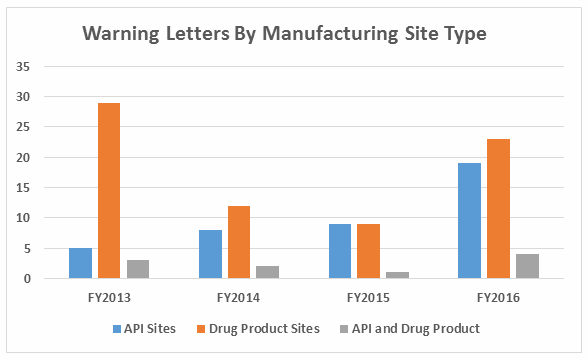
Figure 2
Table 2 shows the geographic distribution of warning letters issued outside the U.S. European countries are counted together and include: Ireland, Spain, Czech Republic, Italy, Portugal, Denmark, Austria, Netherlands, Germany, and Switzerland. The low number of warning letters issued regarding sites in Europe may represent an unofficial reliance on European Medicines Agency (EMA) inspections of sites located in this area, even in the absence of an official mutual recognition agreement (MRA) between the regulatory authorities.
While India received the highest number of warning letters issued to a single country over the four-year time period, China received the most in FY2016 (and second-most over the four years).
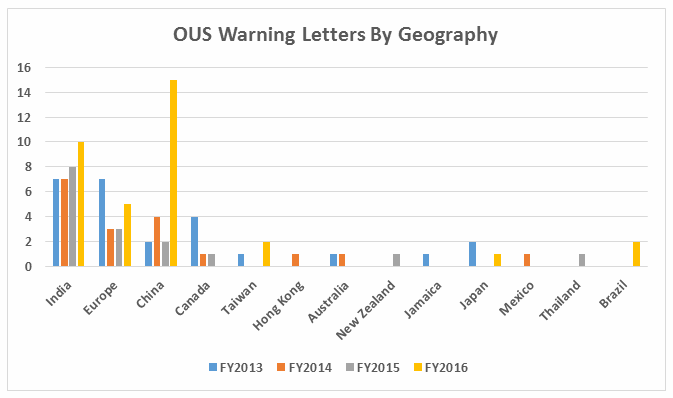
Figure 3
Import Alerts Associated With Warning Letters
Thirty-five warning letters were issued regarding OUS, and 17 of these were associated with import alerts for failure to comply with drug GMPs. So not only did these firms incur the expense associated with remediation of the warning letter, they are prevented from selling product from these sites in the U.S., excluding FDA-identified medically necessary products. Table 3 shows the distribution of the import alerts associated with warning letters in FY2016.
Data Integrity Deficiencies In Warning Letters
Table 4 shows the number of warning letters issued both inside and outside the U.S. that included references to data management and data integrity. This group and analysis excludes those warning letters issued to compounding pharmacies. Data integrity deficiencies in warning letters continue to identify the predicate rule(s) to which firms did not adhere. The percent of warning letters that cite data integrity deficiencies issued to sites in the U.S. is essentially the same as for warning letters issued outside the U.S., though the absolute numbers differ.
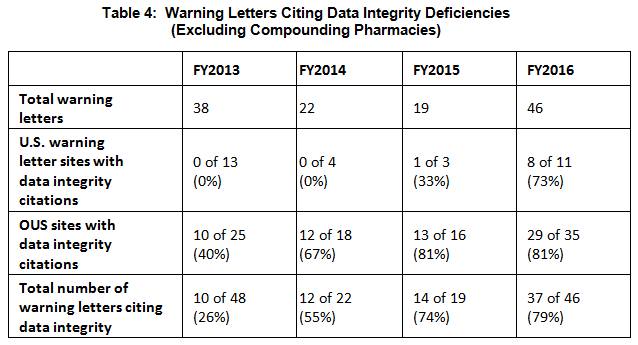
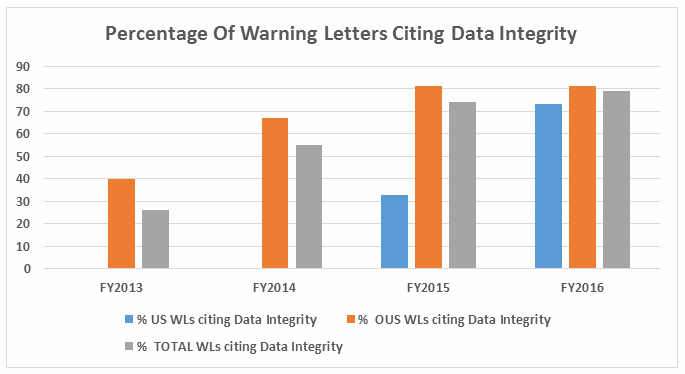
Figure 4
FDA continues to refine the requirements for data integrity remediation that they include in warning letters with the most egregious deficiencies. An example of this may be found at the end of the warning letter issued to Pan Drugs Limited in early 2016 under the heading of Data Integrity Remediation.
Intervals Between Inspections And Warning Letters
Table 5 and Figure 5 show the interval between inspection and issuance of a warning letter over the four fiscal years. The data for the first half of FY2016 is included along with that for the full fiscal year. This interval continues to increase for compounding pharmacies and drug manufacturers the U.S. and remains unchanged for warning letters issued outside the U.S in comparison to FY2015. The only exceptions are for U.S. drug manufacturing sites, excluding compounding pharmacies, in the U.S. in FY2014 and 2015, which saw only 4 and 3 warning letters respectively (see Table 1). (However, we may be seeing a decrease in this interval, which is certainly something to follow in FY2017.) For warning letters issued to sites outside the U.S. in FY2016, Figure 6 compares the intervals between inspection and warning letters and inspection and imposition of an import alert.
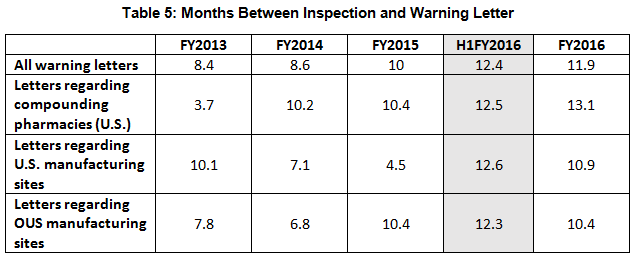
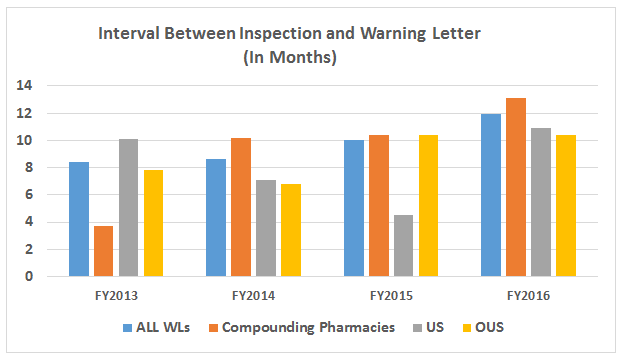
Figure 5
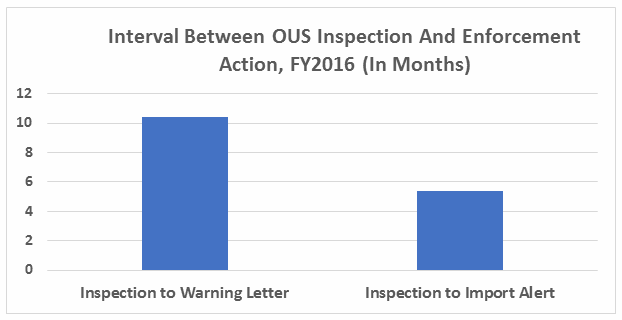
Figure 6
Conclusions
Fiscal year 2016 included both significant changes in drug GMP warning letter enforcement actions and a continued focus on topics we’ve seen for the past three years. FDA issued more than twice the number of drug GMP warning letters in FY2016 than it did in FY2015.
Compounding pharmacies and outsourcing facilities continued to draw disproportionate attention in these enforcement actions, receiving > 50% of all drug GMP warning letters. This focus began in FY2014, after amendments to the Federal Food, Drug, and Cosmetic (FD&C) Act formalized outsourcing facilities in 2013. Outsourcing facilities and compounding pharmacies do not seem to have learned from the previous three years of enforcement in this area — expect continued focus here.
API manufacturing sites, many located outside the U.S., saw an increase in warning letters. Data integrity remains a focus of enforcement actions, and FDA has refined its stated requirements for remediation of data integrity deficiencies.
FY2017 will be an interesting one. Assuming we see a new FDA commissioner, the enforcement focus may change — perhaps diminish — if the incoming administration focuses on decreasing regulations on business. However, I would expect continued enforcement focus on sites outside the U.S., consistent with past years, because that is where most prescription drugs are produced. These actions include issuance of warning letters and imposition of import alerts.
Enforcement actions will continue to be tempered by the need to balance enforcement action with the risk of drug shortages. FDA has also indicated its intent to shorten the time period between inspection and warning letter issuance; we will monitor whether this happens in FY2017. Firms should track current enforcement actions and evaluate any changes in trends as part of a comprehensive GMP intelligence program. This is useful to identify areas for continuous improvement in GMP programs and practices and to minimize surprises during FDA inspections.
About the Author:
 Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. Barbara is currently the co-lead of the Rx-360 Data Integrity Working Group.
Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. Barbara is currently the co-lead of the Rx-360 Data Integrity Working Group.
Before Amgen, Barbara worked for the consulting firm Don Hill and Associates, providing regulatory and quality services to the pharmaceutical industry, and for Eli Lilly and Company in quality and CMC regulatory affairs positions. She began her career in the pharmaceutical / device industry with Hybritech Inc. and received a bachelor's degree in chemistry from the University of Illinois at Urbana-Champaign. You can contact Barbara at bwunger123@gmail.com.
Appendix 1: Drug GMP Warning Letters, FY2016
|
Type |
Date |
Company |
Issued By (District or Center) |
Number of Deficiencies |
Country |
|
Compounding pharmacy |
10/19/2015 |
Kansas City |
5 |
USA |
|
|
Drug product |
10/22/2015 |
Center |
7 |
India |
|
|
Compounding pharmacy |
10/28/2015 |
New England |
6 |
USA |
|
|
Compounding pharmacy |
11/2/2015 |
Atlanta District |
4 |
USA |
|
|
Compounding pharmacy |
11/3/2015 |
Los Angeles |
6 |
USA |
|
|
API and dosage form |
11/5/2015 |
Center |
11 |
India |
|
|
Compounding pharmacy |
11/12/2015 |
New England |
3 |
USA |
|
|
Compounding pharmacy |
12/9/2015 |
San Francisco |
4 |
USA |
|
|
API |
11/25/2015 |
New York |
4 |
USA |
|
|
Dosage form |
12/17/2015 |
Center |
4 |
India |
|
|
Compounding pharmacy |
12/18/2015 |
Cincinnati |
7 |
USA |
|
|
Veterinary drug product |
12/18/2015 |
Kansas City |
4 |
USA |
|
|
Compounding pharmacy |
12/18/2015 |
Detroit |
4 |
USA |
|
|
Compounding pharmacy |
12/22/2015 |
Cincinnati |
4 |
USA |
|
|
API and dosage Form |
12/23/2015 |
Center |
4 |
India (2 sites) |
|
|
Drug product, stem cells |
12/30/2015 |
Center |
4 |
USA |
|
|
API |
12/31/2015 |
Center |
4 |
China |
|
|
API and dosage form |
1/29/2016 |
Center |
4 |
India (3 sites) |
|
|
Compounding pharmacy |
2/1/2016 |
New York |
7 |
USA |
|
|
Compounding pharmacy |
2/3/2016 |
Florida |
4 |
USA |
|
|
Compounding pharmacy |
2/4/2016 |
Florida |
4 |
USA |
|
|
Compounding pharmacy |
2/26/2016 |
Detroit |
4 |
USA |
|
|
Compounding pharmacy |
2/29/2016 |
San Francisco |
5 |
USA |
|
|
Drug product |
3/3/2016 |
Center |
4 |
India |
|
|
Compounding pharmacy |
3/10/2016 |
Florida |
6 |
USA |
|
|
Compounding pharmacy |
3/10/2016 |
Kansas City |
4 |
USA |
|
|
Drug product |
3/10/2016 |
Kansas City |
4 |
USA |
|
|
Compounding pharmacy |
3/14/2016 |
New Jersey |
7 |
USA |
|
|
Compounding pharmacy |
3/16/2016 |
Dallas |
5 |
USA |
|
|
Compounding pharmacy |
3/21/2016 |
New York |
4 |
USA |
|
|
Compounding pharmacy |
3/23/2016 |
Baltimore |
5 |
USA |
|
|
Drug product |
4/1/2016 |
Center |
4 |
India |
|
|
API repackager/relabeler |
4/7/2016 |
New Orleans |
3 |
USA |
|
|
Drug product |
4/12/2016 |
Florida |
7 |
USA |
|
|
API |
4/14/2016 |
Center |
4 |
India |
|
|
Compounding pharmacy |
4/21/2016 |
Los Angeles |
5 |
USA |
|
|
Compounding pharmacy |
4/27/2016 |
New Orleans |
0 |
USA |
|
|
Compounding pharmacy |
4/29/2016 |
Baltimore |
4 |
USA |
|
|
Compounding pharmacy |
5/5/2016 |
Los Angeles |
8 |
USA |
|
|
API |
5/7/2016 |
Center |
4 |
Germany |
|
|
API |
5/12/2016 |
Center |
3 |
China |
|
|
Compounding pharmacy |
5/18/2016 |
New England |
1 |
USA |
|
|
Compounding pharmacy |
5/19/2016 |
Cincinnati |
5 |
USA |
|
|
API |
5/19/2016 |
Center |
3 |
India |
|
|
API and dosage form |
5/20/2016 |
Center |
5 |
Italy |
|
|
Compounding pharmacy |
5/23/2016 |
San Francisco |
7 |
USA |
|
|
Drug product |
5/27/2016 |
Center |
4 |
Taiwan |
|
|
Drug product |
6/2/2016 |
Center |
5 |
Taiwan |
|
|
Compounding pharmacy |
6/3/2016 |
New Orleans |
4 |
USA |
|
|
Compounding pharmacy |
6/3/2016 |
Dallas |
7 |
USA |
|
|
Compounding pharmacy |
6/6/2016 |
New Orleans |
3 |
USA |
|
|
Compounding pharmacy |
6/7/2016 |
New Jersey |
13 |
USA |
|
|
Compounding pharmacy |
6/9/2016 |
New Orleans |
7 |
USA |
|
|
API |
6/16/2016 |
Center |
2 |
China |
|
|
API |
6/21/2016 |
Center |
2 |
China |
|
|
Compounding pharmacy |
6/22/2016 |
New Orleans |
6 |
USA |
|
|
Drug product |
6/22/2016 |
Center |
3 |
China |
|
|
Compounding pharmacy |
6/28/2016 |
Dallas |
7 |
USA |
|
|
API |
6/30/2016 |
Center |
2 |
UK |
|
|
Compounding pharmacy |
7/5/2018 |
New England |
7 |
USA |
|
|
Compounding pharmacy |
7/8/2016 |
Atlanta District |
5 |
USA |
|
|
Drug product, repackager |
7/12/2016 |
Cincinnati |
4 |
USA |
|
|
API |
7/19/2016 |
Center |
3 |
China |
|
|
Compounding pharmacy |
7/22/2016 |
Chicago |
1 |
USA |
|
|
Compounding pharmacy |
7/22/2016 |
Chicago |
4 |
USA |
|
|
Compounding pharmacy |
7/25/2016 |
Dallas |
5 |
USA |
|
|
Drug product |
7/26/2016 |
Center |
5 |
China |
|
|
Compounding pharmacy |
7/29/2016 |
Chicago |
6 |
USA |
|
|
Compounding pharmacy |
8/1/2016 |
New Jersey |
1 |
USA |
|
|
Contract testing laboratory and API |
8/2/2016 |
Los Angeles |
3 |
USA |
|
|
API |
8/4/2016 |
Center |
3 |
China |
|
|
Compounding pharmacy |
8/5/2016 |
New Orleans |
1 |
USA |
|
|
Drug product |
8/5/2016 |
Florida |
5 |
USA |
|
|
Compounding pharmacy |
8/8/2016 |
Baltimore |
1 |
USA |
|
|
Drug product |
8/10/2016 |
Center |
5 |
China |
|
|
API |
8/11/2016 |
Center |
3 |
China |
|
|
Compounding pharmacy |
8/12/2016 |
Baltimore |
77 |
USA |
|
|
API |
8/12/2016 |
Center |
6 |
China (2 sites) |
|
|
Drug product |
8/15/2016 |
Philadelphia |
1 |
USA |
|
|
Compounding pharmacy |
8/15/2016 |
Denver |
1 |
USA |
|
|
Drug product |
8/17/2016 |
PHS Center |
10 |
USA |
|
|
Compounding pharmacy |
8/17/2016 |
Denver |
2 |
USA |
|
|
Compounding pharmacy |
8/18/2016 |
Los Angeles |
7 |
USA |
|
|
API |
8/19/2016 |
Center |
2 |
China (2 sites) |
|
|
Compounding pharmacy |
8/19/2016 |
Dallas |
6 |
USA |
|
|
Drug product |
8/25/2016 |
Center |
4 |
Brazil |
|
|
API |
8/25/2016 |
Center |
4 |
India |
|
|
Compounding pharmacy |
8/25/2016 |
Philadelphia |
4 |
USA |
|
|
Compounding pharmacy |
9/2/2016 |
Chicago |
3 |
USA |
|
|
Compounding pharmacy |
9/2/2016 |
Los Angeles |
11 |
USA |
|
|
API |
9/6/2016 |
Center |
3 |
China |
|
|
Compounding pharmacy |
9/7/2016 |
Seattle |
1 |
USA |
|
|
Drug product |
9/12/2016 |
Center |
4 |
Brazil |
|
|
API |
9/15/2016 |
Center |
3 |
China |
|
|
Drug product |
9/15/2016 |
Center |
3 |
Japan |
|
|
Drug product |
9/26/2016 |
Center |
3 |
Netherlands |
|
|
Compounding pharmacy |
9/20/2016 |
Cincinnati |
4 |
USA |
|
|
Compounding pharmacy |
9/28/2016 |
Los Angeles |
1 |
USA |
|
|
Drug product |
9/29/2016 |
Center |
6 |
Switzerland |
|
|
Drug product |
9/29/2016 |
Center |
2 |
China |
|
|
Compounding pharmacy |
9/30/2016 |
Philadelphia |
7 |
USA |
|
|
Compounding pharmacy |
9/30/2016 |
Chicago |
6 |
USA |