
An Analysis Of 2017 FDA Warning Letters On Data Integrity
By Barbara Unger, Unger Consulting Inc.

This article represents the third year we have published an evaluation of warning letters associated with data governance and data integrity deficiencies (see our 2015 and 2016 editions). Enforcement of failures in data integrity and data governance began almost 20 years ago and continues to increase in visibility and number of warning letter enforcement actions. The FDA is not the only health authority that identifies these issues in inspections and enforcement actions, but the FDA’s transparency ensures the data is available. In this summary, we:
- Briefly review the history that serves as background to where we are now,
- Identify CY2017 warning letters that cite data integrity deficiencies,
- Identify the number of warning letters citing this topic in the past 10 years and the countries where these sites are located, and
- Identify the regulations cited most frequently in CY2017 drug GMP warning letters citing data integrity failures
 Background
Background
Let’s begin with a review of where and when this topic originated. The “generics scandal” of the 1980s identified falsified data submitted to the FDA in support of abbreviated new drug applications (ANDAs). In response, the FDA brought a new focus to pre-approval inspections (PAIs) to evaluate raw laboratory data included in the marketing application and evaluate whether the site was capable of manufacture as described in the application.
In parallel, the FDA recognized the pharmaceutical industry’s increased reliance on computerized systems. In response, the agency developed and published in 1997 21 CFR11, the final rule on Electronic Records and Electronic Signatures and its preamble. While the requirements for electronic signatures were understood, confusion remained on both sides regarding the interpretation and enforcement of requirements for electronic records. Following enforcement actions against Able Laboratories in 2005 and against Ranbaxy in 2006 and 2008, the FDA announced a pilot program in 2010 to evaluate data integrity as part of routine GMP inspections. The FDA planned to use the information gained from these inspections to determine whether revisions to Part 11 or additional guidance on the topic were necessary. FDA investigator Robert Tollefsen describes the program in a slide deck presented at a variety of industry conferences in 2010. In the slide deck, the FDA stresses that it will “continue to enforce all predicate rule requirements, including requirements for records and recordkeeping.” In fact, deficiencies in Part 11 are rarely, if ever, cited in warning letters because almost all failures are those where firms fail to comply with the predicate rules.
Fundamentally, all data integrity deficiencies identified in Form 483s and warning letters are failures to follow CGMPs as specified in the predicate rules. The FDA has not implemented novel interpretations or requirements applicable to data governance. The use of computer systems and other electronic systems requires different approaches to ensure compliant practices, but these are all based on the existing regulations in 21 CFR211.
Data Integrity Drug GMP Warning Letters And Trends From The Past 10 Years
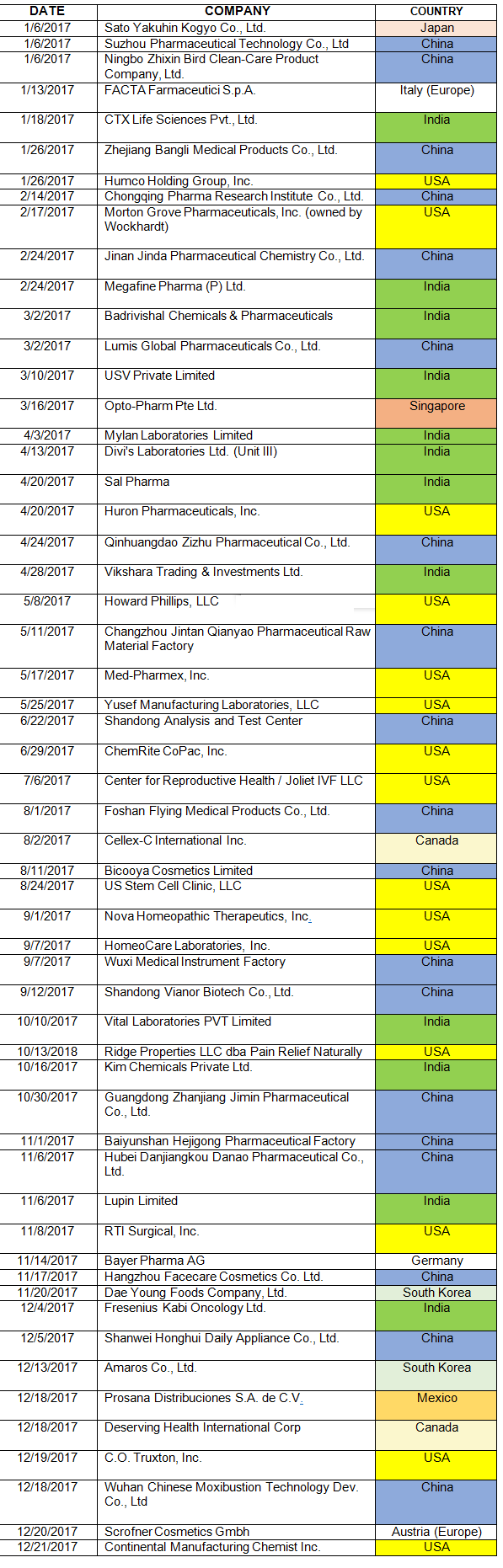
Table 1 lists the warning letters that include data integrity deficiencies, the date of issuance, and the country where the facility is located. The country column is color-coded, and all European countries are consolidated into a single group in subsequent tables and figures. The FDA issued 82 warning letters in CY2017, excluding those issued to compounding pharmacies and outsourcing facilities. Fifty-six included a data integrity component, a total of 68 percent of the warning letters.
Table 1: CY2017 Drug Warning Letters With Data Integrity Deficiencies
As mentioned in the background section, the FDA began enforcement in this area nearly 20 years ago. Table 2 and Figure 2 present data over the last 10 years, CY2008 through CY2017. The number of warning letters including this topic ranged from four to six from 2008 through 2013, doubled in CY2014 to 10, and was followed by a marked increase between CY2015 and the current year. The number of warning letters increased from 15 in 2015 to 41 in 2016 and 56 in 2017. The number of countries associated with these warning letters also increased similarly, and in 2017 nine countries were associated with the sites that were the subject of warning letters.
Table 2: Number of Data Integrity Associated Warning Letters by Country CY2008-2017
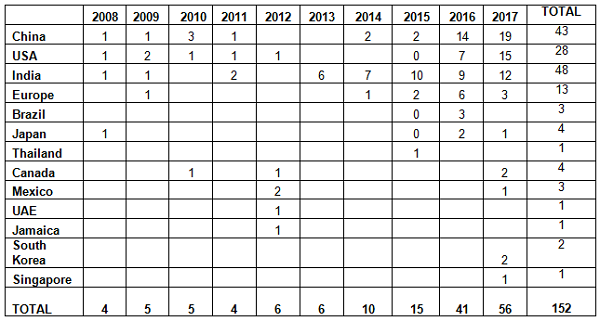
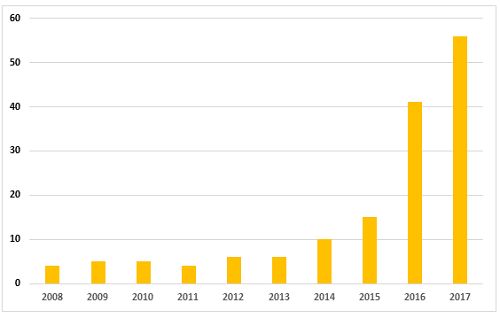
Figure 2: Data integrity associated warning letters, CY2008-CY2017
Table 3 compares the number and percentage of warning letters citing data governance and data integrity in both the past 10 years and the most recent three years. Overall, sites in India have been the subject in the most warning letters of this type, whereas in the past three years, China rose to the head of the list. Overall, the U.S. has received approximately 20 percent of the warning letters, European countries have received approximately 10 percent, and the rest of the world claims approximately 12 percent.
Table 3: Geographic Totals and Percentage, 2015-2017 and 2008-2017
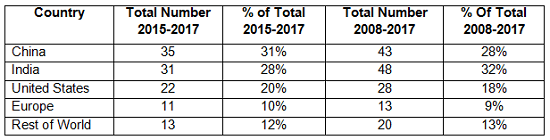
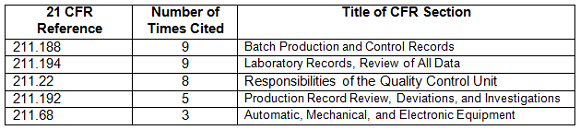
Table 4 shows the regulations most frequently cited in the warning letters in CY2017. Many of the deficiencies did not identify a regulation or are provided by the FDA as “conclusions” or “data integrity remediation” instructions to which the firms must respond. The citation of regulations continues the FDA’s stated goal of focusing on the evaluation of predicate rule requirements.
Table 4: Regulations Cited in 2017 Data Integrity Associated Drug Warning Letters
Actions Firms Can Take To Prevent, Identify, And Remediate Issues
The FDA initiated enforcement actions in this area as early as 1999 and continued to the point where the last three years have seen data integrity cited in 68 to 80 percent of warning letters. We have seen ever-increasing participation by global health authorities.
So, what is a firm to do to prevent, detect, and remediate these problems before the health authorities become involved? We divide these actions into ones that may be taken by executive management and functional areas. This year, we include a focus on management of contract services among the actions for firms to consider. Additional detail on contract manufacture and data governance is provided in two articles published in 2017. Find them HERE and HERE.
Executive Management Ownership
- Executive management must understand that health authority focus on this area is not going to diminish. Data governance applies to both paper and electronic data throughout its life cycle.
- Executive management must establish and maintain a corporate culture of openness where employees may report problems and failures without fear of retribution. In fact, reporting of problems should be encouraged and rewarded.
- Executive management must own the gap assessment process and remediation efforts. Remediation may be costly and time-consuming. Firms often uncover additional problems along the way. Don’t expect to complete remediation quickly; it’s often a multiyear process.
Be ready to respond if you should receive a Warning Letter. Learn how in the course:
Writing Effective 483 and Warning Letter Responses
Technical Area Actions
- Cross-functional teams should perform gap assessments for both paper and computer systems against predicate rule requirements and specific data governance/integrity guidance from health authorities. The team should identify corrective actions and a timeline for their implementation. Firms should implement interim corrective actions until they can put fully compliant solutions in place.
- Firms should map data and process flows and identify and remediate risk areas. Results from this exercise can contribute to the gap assessments described above.
- Firms should validate systems for their intended purpose and ensure that adequate controls are in place to ensure that deleted or altered data can be detected.
- Monitor enforcement actions including Form 483s, warning letters, import alerts, EU reports of GMP noncompliance, and WHO Notices of Concern. All of these, except for the Form 483s, are available without cost on the internet, and Form 483s are available from commercial sources.
- Ensure that the data governance processes at suppliers and contract service providers are adequate to ensure that data is valid and trustworthy. This effort begins with rigorous due diligence evaluations, periodic on-site oversight, and appropriately detailed quality agreements.
Conclusion
Data integrity and data governance remain initiatives of global health authorities. The U.K.’s Medicines and Healthcare Products Regulatory Agency (MHRA) was the earliest to enter the area, in 2015, with its guidance and a published draft revision in 2016. The European Medicines Agency (EMA), World Health Organization (WHO), Pharmaceutical Inspection Co-operation Scheme (PIC/S), Australia, Canada, and China followed in 2016. Further, this is not limited to the GMP area but now includes good clinical practice (GCP), with the most impactful cases at sites that perform bioavailability and bioequivalence studies. For these firms, the data for hundreds of products is impacted. Sponsors must frequently repeat the studies at different sites. Most recently, this has included failures identified at GVK and Semler Research. Consequences at Semler included a three-page Form 483, untitled letter, WHO notice of concern, and EMA recommendation of suspension.
GMP enforcement citing data governance and data integrity has not diminished, expanding both the number of warning letters and their geographic distribution. Although the number of warning letters has increased markedly over the past three years, the percentage has decreased slightly. In CY2017 an increasing number of countries were home to sites that were the subject of these warning letters. Deficiencies in data governance and data integrity have remained markedly consistent over the 10 years addressed in this report, with a few new areas identified each year. This year saw the addition of three new focus areas, including:
- Firms that repackage APIs were transferring analytical results onto a Certificate of Analysis on their letterhead, making it appear that they generated the results. The practice obscures the supply chain from the company that purchases and uses the material in the manufacture of drug products.
- Firms aborted an excessive number of analytical of runs.
- Firms manipulated “integration suppression” parameters within chromatography data systems, intending to obscure or minimize impurity peaks.
These three areas merit our attention as we progress through 2018. I expect this type of problem to expand in scope to more OTC manufacturers because actions in this area is a clear trend that began in 2017. I also watch for this topic to be cited more frequently in enforcement actions taken against compounding pharmacies and outsourcing facilities. Previously, most of the problems in this area addressed failures in aseptic processing, including facilities and equipment issues. I look for data integrity to be cited more frequently in both Form 483s and warning letters issued to these firms.
Note: Readers who want the complete text of the warning letter deficiencies on this topic can find them on my website for 2015 (starting on page 4), 2016 (starting on page 5), and 2017 (starting on page 8).
About The Author:
 Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP-related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP- and supply chain-related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. She is currently the co-lead of the Rx-360 Data Integrity Working Group.
Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP-related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP- and supply chain-related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. She is currently the co-lead of the Rx-360 Data Integrity Working Group.
Unger received a bachelor's degree in chemistry from the University of Illinois at Urbana-Champaign. You can contact her at bwunger123@gmail.com.