
An Analysis Of MHRA's Annual GMP Inspection Deficiencies Report
By Barbara Unger, Unger Consulting Inc.
The U.K.’s Medicines and Healthcare products Regulatory Agency (MHRA) has taken a different approach in the publication of the GMP deficiencies for drug product issued during inspections in 2018 and published in October 2019. In 2015 and 2016, the MHRA provided approximately 100 slide decks with tables, figures, and text from deficiencies against the requirements in the chapters and annexes. No data were published for 2017. The MHRA published a 6,200+ line Excel spreadsheet of its 2018 GMP inspection data so that individuals can parse and present the data according to their needs.
publication of the GMP deficiencies for drug product issued during inspections in 2018 and published in October 2019. In 2015 and 2016, the MHRA provided approximately 100 slide decks with tables, figures, and text from deficiencies against the requirements in the chapters and annexes. No data were published for 2017. The MHRA published a 6,200+ line Excel spreadsheet of its 2018 GMP inspection data so that individuals can parse and present the data according to their needs.
This two-part article is data dense and includes many tables and figures. Part 1 begins with a high-level overview of the 2018 data, including additional trends from the two most recent MHRA reports, from 2015 and 2016. It then goes on to identify and evaluate the critical and major deficiencies from 2018, identifying the chapters and annexes associated with critical and major deficiencies. For critical deficiencies citing Chapter 1 and Annex 1, the specific paragraphs and requirements with which they are associated are identified.
Part 2 presents the 10 most frequently cited paragraphs for the 10 chapters and annexes with the most frequently cited deficiencies identified in Part 1.
Background
I found the “Notes and Guidance” segment of the MHRA spreadsheet a bit confusing. In light of this, here’s how I’ve parsed the data. Each row in the spreadsheet is treated as a unique deficiency, regardless of whether it is a “deficiency” or a “sub-point” as identified in the Notes and Guidance section of the MHRA publication. I cannot discern which is which in the spreadsheet, so I treat them all equally. The summary data from 2015 and 2016 are taken directly from the MHRA 2016 report. Data were not posted for 2017. Any mistakes in this analysis and reporting of the 2018 data are mine, not the MHRA’s.
Overall Data
The MHRA conducted 285 GMP inspections, both domestic and outside of the U.K., in 2018. Inspections outside the U.K. were conducted in Bangladesh, China, India, Japan, Singapore, South Korea, and the United States. Country-specific data, other than that provided for the U.K. and “overseas,” was not provided in the 2015 and 2016 MHRA reports. The 285 inspections from 2018 reflect a decrease from the number of inspections conducted in both 2015 and 2016.
Table 1 identifies the number of drug product inspections, by country, performed by the MHRA in 2018. As in past years, almost all MHRa inspections were conducted in the U.K. The percentage of inspections conducted in the U.K. in 2018 increased slightly from 2015 and 2016 and the percentage of overseas inspection decreased slightly in 2018 to 20 percent of the total.
In 2018, the largest number of inspections conducted outside the U.K. were performed in India, with a total of 43. The remaining 14 inspections outside the U.K. were conducted in six countries, with China and the U.S. each the subject of five inspections, and the other countries with one each.
Table 1: MHRA Inspections by Country
|
Country |
Number of Inspections 2015 / % total |
Number of Inspections 2016 / % total |
Number of Inspections 2018 / % total |
|
Total |
303 |
324 |
285 |
|
U.K. |
224 / 74% |
242 / 75% |
228 / 80% |
|
Overseas Inspections |
79 / 26% |
82 / 25% |
57 / 20% |
|
India |
|
|
43 / 15% |
|
China |
|
|
5 / 2% |
|
United States |
|
|
5 / 2% |
|
Bangladesh |
|
|
1 / 0.3% |
|
South Korea |
|
|
1 / 0.3% |
|
Singapore |
|
|
1 / 0.3% |
|
Japan |
|
|
1 / 0.3% |
Table 2 shows the trend, including all classification of deficiencies from 2015, 2016, and 2018 identified in the top 10 chapters and annexes. Data from 2015 and 2016 are taken directly from the 2016 report published by the MHRA. Quality Systems leads the list in all three years. Notable changes in 2018 from the two previous years include:
- Outsourced Activities (Chapter 7) appears within the top 10 in 2018, though it did not appear in either of the two other years.
- Personnel (Chapter 2) is no longer among the top 10 in 2018.
- Computerised Systems (Annex 11) remains within the top 10 in 2018, although it fell from fifth place in 2015, to seventh in 2016, and to 10th place in 2018.
- Complaints and Recalls (Chapter 8) also diminished in rank over the time period, from second in 2015, to fourth in 2016, and eighth in 2018.
Table 2: Overall Deficiency Trend Comparison, Top 10
|
Rank |
2015 |
2016 |
2018 |
|
1 |
Quality Systems |
Quality System |
Quality System |
|
2 |
Complaints and Recalls |
Sterility Assurance |
Documentation |
|
3 |
Documentation |
Production |
Production |
|
4 |
Quality Control |
Complaints and Recall |
Validation / Qualification |
|
5 |
Computerised Systems |
Qualification / Validation |
Premises and Equipment |
|
6 |
Production |
Premises and Equipment |
Sterility Assurance |
|
7 |
Premises and Equipment |
Computerised Systems |
Quality Control |
|
8 |
Validation |
Personnel |
Complaints and Recall |
|
9 |
Personnel |
Documentation |
Outsourced Activities |
|
10 |
Materials Management |
Quality Control |
Computerised Systems |
Figure 1 and Figure 2 present the total number of deficiencies according to the GMP chapter or GMP annex cited, respectively. These figures include all deficiency classifications — critical, major, and other. Annex 15, Qualification and Validation, and Annex 1, Sterility Assurance, take first and second place among the most frequently cited annexes, with approximately 600 and 450 deficiencies, respectively. This is followed by Annex 11, Computerized Systems, and Annex 16, Certification by a Qualified Person and Batch Release. All other annexes are associated with double-digit or fewer deficiencies.
Chapter 1, Quality Management, with approximately 1,500 citations, has more than twice the number of deficiency citations as the nearest chapter. Chapter 4, Documentation, with almost 760 deficiencies, is closely followed by Chapter 5, Production, with just over 650, and Chapter 3, Premises and Equipment, at just under 570 citations. Chapters 2, 6, 7, and 8 each had between 200 and 400 deficiencies and, of the two remaining two chapters, one had fewer than 50 citations and the other had just one citation.
Figure 3 shows the top 10 categories when values from annexes and chapters are combined; the same information is provided in Table 2 with the number of associated deficiencies. Again, this includes all categories of deficiencies. The top 10 citations in each of these categories will be identified in Part 2.
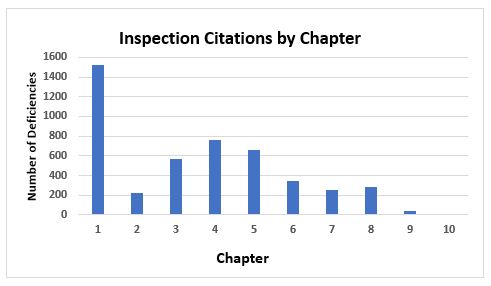
Figure 1: Inspection citation by GMP chapter
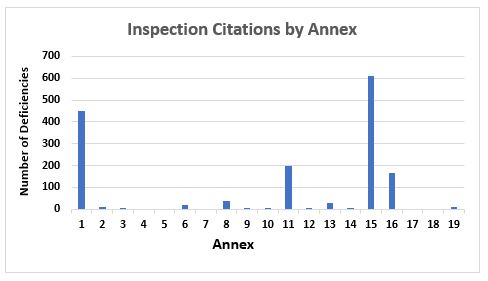
Figure 2: Inspection citations by GMP annex
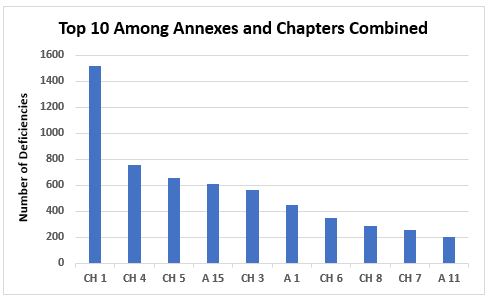
Figure 3: Top 10 chapters and annexes cited in deficiencies
Critical And Major Deficiencies
Table 3 provides a tabulation of all 2018 deficiencies by their classification. Critical deficiencies clearly constitute the smallest category; additional detail on these will be provided later. Major deficiencies constitute almost 40 percent of the total, and other deficiencies constitute the majority of the deficiencies at almost 60 percent of the total.
Table 3: Number of Deficiencies by Classification in 2018
|
Classification |
Number |
Percentage of Total |
|
Critical |
142 |
2 |
|
Major |
2391 |
39 |
|
Other |
3676 |
59 |
The majority of critical and major deficiencies among the 6,200-plus deficiencies cluster in a few chapters and annexes. Figure 4 shows the number of critical deficiencies and the chapters or annexes that are referenced. Among the critical deficiencies, 37 percent are associated with Chapter 1, Quality Systems, and 22 percent are associated with Annex 1, Sterility Assurance. Annexes 1 and 11 are the only other annexes that include citations for critical deficiencies; the remainder cite GMP chapters.
Tables 4 and 5 provide the specifics on the citations for Chapter 1 and Annex 1, respectively. The tables include the identifying paragraph, the number of times the deficiency was cited, and a short version of the text in the GMP guide.
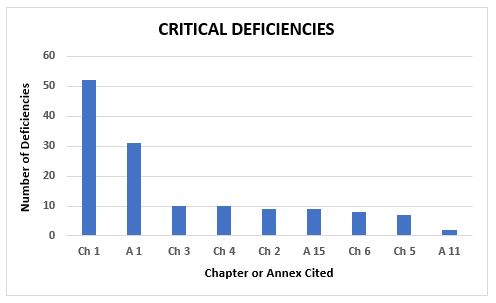
Figure 4: Critical deficiencies
Table 4: Critical Deficiencies Cited in Chapter 1
|
Critical Deficiencies |
||
|
Paragraph |
# |
Short Version of Text |
|
1.4(xiv) |
9 |
An appropriate level of root cause analysis should be applied during the investigation of deviations, suspected product defects and other problems. This can be determined using Quality Risk Management principles… |
|
1.4(viii) |
6 |
A state of control is established and maintained by developing and using effective monitoring and control systems for process performance and product quality. |
|
1.8(vii) |
6 |
Any significant deviations are fully recorded, investigated with the objective of determining the root cause and appropriate corrective and preventive action implemented; |
|
1.8(vi) |
4 |
Records are made, manually and/or by recording instruments, during manufacture which demonstrate that all the steps required by the defined procedures and instructions were in fact taken and that the quantity and quality of the product was as expected. |
|
1 Principle |
4 |
The holder of a Manufacturing Authorisation must manufacture medicinal products so as to ensure that they are fit for their intended use, comply with the requirements of the Marketing Authorisation or Clinical Trial Authorisation, as appropriate and do not place patients at risk due to inadequate safety, quality or efficacy… |
|
1.5 |
3 |
Senior management has the ultimate responsibility to ensure an effective Pharmaceutical Quality System is in place, adequately resourced and that roles, responsibilities, and authorities are defined, communicated and implemented throughout the organisation… |
|
1.4(i) |
2 |
Product realisation is achieved by designing, planning, implementing, maintaining and continuously improving a system that allows the consistent delivery of products with appropriate quality attributes. |
|
1.4(xii) |
2 |
Arrangements are in place for the prospective evaluation of planned changes and their approval prior to implementation taking into account regulatory notification and approval where required. |
|
1.8(iii) |
2 |
All necessary facilities for GMP are provided including… |
|
1.8(xi) |
2 |
Complaints about products are examined, the causes of quality defects investigated and appropriate measures taken in respect of the defective products and to prevent reoccurrence. |
|
1.3 |
1 |
The size and complexity of the company’s activities should be taken into consideration when developing a new Pharmaceutical Quality System or modifying an existing one… |
|
1.1 |
1 |
Quality Management is a wide-ranging concept, which covers all matters, which individually or collectively influence the quality of a product… |
|
1.12 |
1 |
Quality risk management is a systematic process for the assessment, control, communication and review of risks to the quality of the medicinal product. It can be applied both proactively and retrospectively. |
|
1.4(xi) |
1 |
Continual improvement is facilitated through the implementation of quality improvements appropriate to the current level of process and product knowledge. |
|
1.4(xiii) |
1 |
After implementation of any change, an evaluation is undertaken to confirm the quality objectives were achieved and that there was no unintended deleterious impact on product quality. |
|
1.6 |
1 |
There should be periodic management review, with the involvement of senior management, of the operation of the Pharmaceutical Quality System to identify opportunities for continual improvement of products, processes and the system itself. |
|
C1.8(i) |
1 |
All manufacturing processes are clearly defined, systematically reviewed in the light of experience and shown to be capable of consistently manufacturing medicinal products of the required quality and complying with their specifications. |
|
C1.8(v) |
1 |
Procedures are carried out correctly and operators are trained to do so; |
|
C1.8(x) |
1 |
A system is available to recall any batch of product, from sale or supply; |
|
C1.9(iii) |
1 |
Test methods are validated; |
|
C1.9(iv) |
1 |
Records are made, manually and/or by recording instruments, which demonstrate that all the required sampling, inspecting and testing procedures were actually carried out… |
|
C1.9(i-viii) |
1 |
Quality Control is that part of Good Manufacturing Practice which is concerned with sampling, specifications and testing, and with the organisation, documentation and release procedures which ensure that the necessary and relevant tests are actually carried out and that materials are not released for use, nor products released for sale or supply, until their quality has been judged to be satisfactory… |
Table 5: Critical Deficiencies Cited in Annex 1
|
Critical Deficiencies |
||
|
Paragraph |
# |
Short Version of Text |
|
A1.18 |
3 |
Where aseptic operations are performed monitoring should be frequent using methods such as settle plates, volumetric air and surface sampling (e.g. swabs and contact plates)… |
|
A1.20 |
3 |
Appropriate alert and action limits should be set for the results of particulate and microbiological monitoring. If these limits are exceeded operating procedures should prescribe corrective action. |
|
A1.19 |
2 |
Recommended limits for microbiological monitoring of clean areas during operation:… |
|
A1.46 |
2 |
In clean areas, all exposed surfaces should be smooth, impervious and unbroken in order to minimize the shedding or accumulation of particles or micro-organisms and to permit the repeated application of cleaning agents, and disinfectants where used. |
|
A1.51 |
2 |
Changing rooms should be designed as airlocks and used to provide physical separation of the different stages of changing and so minimize microbial and particulate contamination of protective clothing… |
|
A1.52 |
2 |
Both airlock doors should not be opened simultaneously. An interlocking system or a visual and/or audible warning system should be operated to prevent the opening of more than one door at a time. |
|
A1.73 |
2 |
Activities in clean areas and especially when aseptic operations are in progress should be kept to a minimum and movement of personnel should be controlled and methodical, to avoid excessive shedding of particles and organisms due to over-vigorous activity… |
|
A1.1 |
1 |
The manufacture of sterile products should be carried out in clean areas entry to which should be through airlocks for personnel and/or for equipment and materials… |
|
A1.21 |
1 |
The utilisation of isolator technology to minimize human interventions in processing areas may result in a significant decrease in the risk of microbiological contamination of aseptically manufactured products from the environment… |
|
A1.3 |
1 |
Clean areas for the manufacture of sterile products are classified according to the required characteristics of the environment… |
|
A1.33 |
1 |
Handling and filling of aseptically prepared products should be done in a grade A environment with a grade B background. |
|
A1.37 |
1 |
All personnel (including those concerned with cleaning and maintenance) employed in such areas should receive regular training in disciplines relevant to the correct manufacture of sterile products… |
|
A1.40 |
1 |
Wristwatches, make-up and jewelry should not be worn in clean areas. |
|
A1.41 |
1 |
Changing and washing should follow a written procedure designed to minimize contamination of clean area clothing or carry-through of contaminants to the clean areas. |
|
A1.44 |
1 |
Outdoor clothing should not be brought into changing rooms leading to grade B and C rooms. |
|
A1.45 |
1 |
Clean area clothing should be cleaned and handled in such a way that it does not gather additional contaminants which can later be shed… |
|
A1.49 |
1 |
Pipes and ducts and other utilities should be installed so that they do not create recesses, unsealed openings and surfaces which are difficult to clean. |
|
A1.53 |
1 |
A filtered air supply should maintain a positive pressure and an air flow relative to surrounding areas of a lower grade under all operational conditions and should flush the area effectively… |
|
A1.55 |
1 |
A warning system should be provided to indicate failure in the air supply. Indicators of pressure differences should be fitted between areas where these differences are important… |
|
A1.62 |
1 |
Disinfectants and detergents should be monitored for microbial contamination; dilutions should be kept in previously cleaned containers and should only be stored for defined periods unless sterilised… |
|
A1.64 |
1 |
Precautions to minimize contamination should be taken during all processing stages including the stages before sterilisation. |
|
A1.85 |
1 |
For effective sterilisation the whole of the material must be subjected to the required treatment and the process should be designed to ensure that this is achieved. |
Figure 5 shows 12 annexes and chapters associated with major deficiencies and the number of times they were cited. In addition to these 12 chapters and annexes, major deficiencies were identified in Chapter 9 (11), Annex 2 (4), Annex 8 (8), Annex 3 (3), Annex 13 (3), and one each in Annex 6 and Annex 19. More GMP chapters and annexes were cited in major deficiencies than were cited in critical deficiencies. Among the major deficiencies, 33 percent are associated with Chapter 1, 11 percent are associated with Annex 15, 11 percent are associated with Annex 1, and 10 percent are associated with Chapter 5. Shortcomings in Quality Systems clearly lead the list of both critical and major deficiencies, demonstrating the agency’s focus on the importance of a sound quality system to GMP compliance.
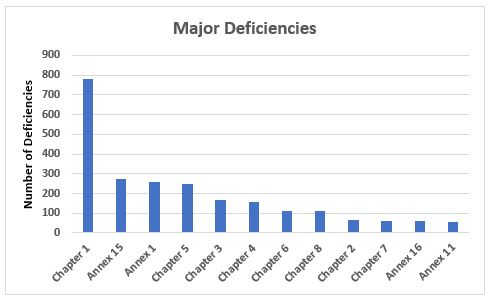
Figure 5: Major deficiencies
Conclusions
- In 2018 the MHRA conducted fewer inspections than in 2015 and 2016. The percentage of inspections conducted in the U.K. increased slightly and those outside the U.K. decreased slightly over that period. It will be useful to monitor whether the MRA with the FDA results in a decrease in the number of inspections of sites that they may both inspect. Because the MHRA only conducted five inspections in the U.S., it will be difficult to determine if the MRA with the FDA results in a decrease of inspection in the U.S.
- Chapter 7, Outsourced Activities, was new among the top 10 in 2018 but was not in the top 10 in either 2015 or 2016, and Chapter 2, Personnel, was no longer in the top 10 in 2018.
- In no surprise that Quality Systems, Chapter 1, continues to be first among the areas cited in inspection deficiencies, regardless of classification, with more than double the number of deficiencies of the next area, Chapter 4. Chapter 1 is first among all deficiencies regardless of classification, first in number among the critical deficiencies, and first in number among the major deficiencies.
- Critical deficiencies constitute only 2 percent of the total identified in 2018 and these are associated primarily with Chapter 1 and Annex 1. Approximately 37 percent of critical deficiencies cite requirements in Chapter 1 and approximately 22 percent cite requirements in Annex 1.
- Among the major deficiencies that constitute approximately 40 percent of the total deficiencies, Chapter 1 again leads the group with almost 800 deficiencies. The next three include Annex 15 with approximately 270, Annex 1 with approximately 260, and Chapter 5 with approximately 250. Clearly, Quality Systems is again the substantial leader, as it is for critical deficiencies. Chapter 7, Chapter 8, and Annex 16 were among the top 10 in major deficiencies yet not among the top 10 for critical deficiencies.
- Computerised Systems, Annex 11, remains in the top 10, reinforcing the importance of this area to data integrity and the regulator’s focus on the control and management of electronic data.
Hopefully, the MHRA will publish similar data in 2019 to which we can compare these data. It would be useful for the MHRA to publish the actual text of deficiencies from the various areas in future years as it did in the past. But in the absence of that, publication of these data is valuable and appreciated by the industry.
Acknowledgements:
I want to acknowledge the kind assistance provided by Eileen Counihan, Zachary Unger, and Clare Nolan in helping me learn and negotiate the pivot tables that made this article possible.
About The Author:
 Barbara Unger formed Unger Consulting, Inc. in 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She was co-lead of the Rx-360 Data Integrity Working Group from 2017–2019 and remains a group member. You can contact her at bwunger123@gmail.com.
Barbara Unger formed Unger Consulting, Inc. in 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She was co-lead of the Rx-360 Data Integrity Working Group from 2017–2019 and remains a group member. You can contact her at bwunger123@gmail.com.