
Are You Prepared For The EU's FMD Safety Features Delegated Regulation?
By Eamonn O’Mathuna, Enterprise System Partners (ESP)

The European Union’s (EU) Falsified Medicines (FMD) Directive 2011/62/EU amends Directive 2001/83/EC to safeguard public health by protecting the pharmaceutical supply chain from infiltration by falsified (or counterfeit) medicines. It also introduces new rules to more rigorously regulate the supply chain.
The Commission Delegated Regulation (EU) 2016/161 supplements Directive 2001/83/EC with detailed rules regarding safety features for the packaging of medicinal products for human use. The regulation was adopted in October 2015 and published in the Official Journal of the European Union in February 2016, and it will come into force on February 9, 2019 (three years after publication).
This directive will impact the entire pharmaceutical supply chain, from drug substance manufacturers to pharmacists dispensing medicines at point of sale. This article will provide an explanation of the directive, its impact on the distribution of medicines, and the latest update affecting pharmaceutical manufacturers.
Scope Of The Delegated Regulation
The delegated regulation applies to saleable units of all prescription only medicines (POMs), except for products on the whitelist (e.g., medicinal gases and radionuclide generators). Over–the-counter (OTC) drugs are generally not affected, except for those on the blacklist (i.e., at risk of falsification).
The delegated regulation applies to all 28 countries in the EU and also includes the European Free Trade Association (EFTA) countries (Switzerland, Lichtenstein, Norway, and Iceland). Three countries have received derogations to extend the compliance deadline to nine years (2025): Belgium, Greece, and Italy. At the time of writing, Belgium had indicated that they will not use this extension but instead will go live in 2019.
The main components of the delegated regulation are:
- Safety features
- End–to-end verification model
- Verification of high-risk practices within the supply chain.
Safety Features
The delegated regulation calls for two distinct safety features, both of which need to be added to saleable units to enhance product security. These are a unique identifier (UI) and a tamper-evident device.
The legislation defines rules for the addition of the UI (commonly known as serialization), for product coding, and for the upload of data to European repositories or databases.
On the other hand, the legislation provides less detail on tamper-evidence devices, other than to make their addition mandatory. The European Committee for Standardization (CEN) standard 16679:2014 Packaging – Tamper Verification Features for Medicinal Product Packaging documents industry best practices for tamper-evidence devices.
In general the UI will consist of the following data elements, which will be printed in human readable form and also encoded in a 2D matrix:
- Product code (GTIN, NTIN, or PPN)
- Randomized serial number
- National Health Reimbursement Number (if required by destination country)
- Expiration date
- Batch or lot number

End-To-End Verification Model
The end-to-end serialization model requires the manufacturer to print the UI on each saleable unit and then upload that UI to European repositories at one end of the supply chain. Then, at the other end — the point of dispense of the product — the UI and the tamper evidence device are verified by the pharmacist against the European repositories. If the UI is found to be verified correctly, it is then dispensed and turned off in the repository.
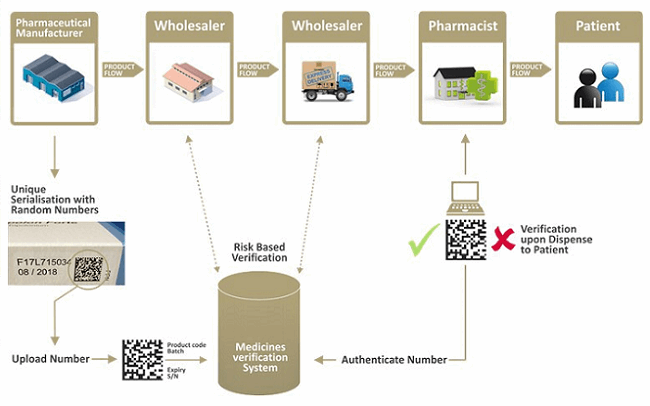
Source: EMVO European Medicines Verification System 2017
To support this model, a collection of national repositories called National Medicine Verification Systems (NMVS) will be connected together via a centralized European Medicine Verification System (EMVS). Manufacturers may upload data to the central EMVS when the saleable units are released for sale. The EMVS will then distribute the UI to the appropriate NMVSs. At the point of dispense, the pharmacy system will then verify the UI against the NMVS in its market.
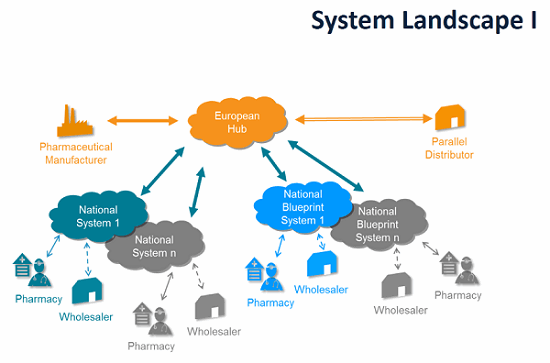
Source: EMVO European Medicines Verification System 2017
Verification Of High-Risk Practices Within The Supply Chain
In addition to verification of the UI at the point of dispense, the delegated regulation also identifies certain high-risk (of falsification) processes that need to be enhanced.
For example, product returned to a wholesaler for resale must, in the future, be verified to still be valid in the European repositories. Product received from a source that is not designated by the manufacturer must also be verified.
Secondly, product that is not destined for sale in the EU must be verified and turned off in the European repositories. Examples include: samples to regulatory agencies, EU product shipped to markets outside the EU, product for destruction, etc.
In these instances, the verification process must include checking both the UI and the tamper-evident device.
Regulatory Information
Unfortunately pharmaceutical processes in Europe are not always harmonized. This is true also for FMD serialization requirements. Each market is free to interpret the delegated regulations and determine how to implement them.
For example, many markets — Germany, Austria, Belgium, Portugal, and the Nordics, to name a few — already have existing pharmaceutical item number systems that are used extensively within their supply chains. As markets decide how to implement the FMD serialisation model in their local markets, usage of these legacy product codes will need to change. It is therefore very important that any changes in these areas are communicated well in advance, so that supply chain partners can adapt their system to meet the local requirements.
Multi-Market Packs
Multi-market packs are manufactured with the intent to be sold in several markets. The European repositories have been designed to support these packs, and as the individual market decide on the implementation rules we may find packs with several product codes on them. For example, a pack could potentially bear the NHRN (National Healthcare Reimbursement Number) for identification in one market and the GTIN (Global Trade Item Number) in another market.
Another consideration for multi-market packs is when the intended destination countries will go live with the new system. For example, if Cyprus, shares packs with Greece, the UIs will be uploaded to EMVS when the packs are released, but when the packs are shipped to Greece they must be turned off in the EMVS, since Greece will be outside the EMVS system until 2025. The same applies to Italy.
EMVS Upgrade Imminent
This summer, the EMVS implemented a system upgrade to bring new features and enhancements online. One of the main enhancements is the expansion of the product master data to be uploaded. The goal is to align the product master data with the different information needs for multiple markets presentations and with the European Identification of Medicinal Products (IDMP) project. It is highly recommended that, once this upgrade has been completed, manufacturers immediately complete the onboarding process, collect master data, and upload this product master data — so that they are very familiar with the process as the FMD deadline approaches.
About the Author:
 Eamonn O’Mathuna is a manufacturing systems consultant with Enterprise System Partners (ESP), with particular expertise in manufacturing execution systems (MES), automation, and serialization. He has over 30 years’ experience as a manufacturing systems expert in Big Pharma, with over 10 years’ niche focus on serialization. During this time, he played a key role as technical lead on a 2007 serialization site rollout for a multinational biopharmaceutical company. Since joining ESP in 2012, Eamonn has played a pivotal role on several manufacturing site and supply chain serialization projects.
Eamonn O’Mathuna is a manufacturing systems consultant with Enterprise System Partners (ESP), with particular expertise in manufacturing execution systems (MES), automation, and serialization. He has over 30 years’ experience as a manufacturing systems expert in Big Pharma, with over 10 years’ niche focus on serialization. During this time, he played a key role as technical lead on a 2007 serialization site rollout for a multinational biopharmaceutical company. Since joining ESP in 2012, Eamonn has played a pivotal role on several manufacturing site and supply chain serialization projects.