
Balancing Speed-to-Market With A Risk-based Approach To Compliance

By Warren Perry, GRCP, Dassault Systèmes BIOVIA Compliance Consultant
Bringing a new drug to market requires significant time – roughly 12 years on average in the U.S., where “only five in 5,000 drugs that enter preclinical testing progress to human testing. One of these five drugs that are tested in people is [eventually] approved. The chance for a new drug to actually make it to market is thus only one in 5,000,” according to Medicine.Net. (1)
Add to these slim odds the increasing costs of meeting compliance standards, and the sum is the newest challenge R&D laboratories face. Addressing compliance pressures can drastically affect productivity and manufacturing cycles, so a risk-based approach to compliance can help biopharmaceutical companies balance the scales. When not everyone in the industry can be “first-to-market,” it’s important to glean information from being the “first-to-fail.” And if a new drug is not going to make it to market, companies will benefit from knowing that sooner rather than later and cutting their financial losses earlier.
FDAReview.org reports that, “In 1980, the typical drug underwent 30 clinical trials involving about 1,500 patients. By the mid-1990s, the typical drug had to undergo more than 60 clinical trials involving nearly 5,000 patients.” (2) With increased regulations and the enormous workload placed on the FDA, the burden lies on biopharmaceutical companies engaged in research to increase efficiencies within their entire development process, helping to create products more quickly and safely by taking a risk-based approach to compliance.
The speedier the research, the better the outcome for everyone. For this reason companies should maintain records from their discovery phases in hopes they may be relevant for the next projects their scientists work on. There’s no need to repeat valid science and good research that simply leads to disappointing conclusions for a certain drug candidate. To be useful later, it’s important to keep all records in a controlled, compliant manner so future researchers can readily access and use them.
Closed Loop Compliance
Balancing quality compliance and speed-to-market goals requires an enterprise-wide solution to help take control of quality commitments throughout typical company “silos” that often hamper productivity. These departments usually include quality, safety, regulatory, clinical, non-clinical, and manufacturing, and they often house data and documents in disparate systems that aren’t easily accessed by others – intentionally or unintentionally. Migrating to a model in which integrated digital systems are in place enables a closed-loop process that’s essential.
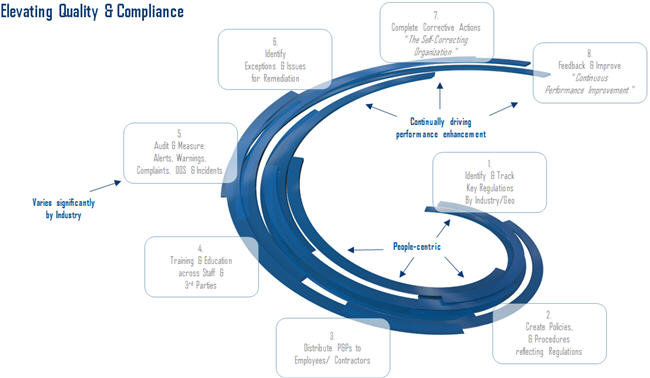
As Figure 1 illustrates, closed loop compliance functions include these steps:
- Identifying key regulations
- Documenting policies and procedures
- Distributing and notifying teams
- Acknowledging information was “read and understood”
- Recording audit and measurement activities
- Identifying exceptions
- Completing corrective actions
- Capturing feedback and improvement procedures
…and then, of course, repeating the cycle. The right connectivity throughout departments helps with accountability and ensures data resides in a system rather than with individuals who might eventually leave the company.
We know piles of data are generated and accumulated throughout research and product lifecycles – from test results, deviation lists, and quality metrics, to out-of-specification reports from the lab and lot release results, to name just a few. Preserving this information for reuse brings quality to the entire process in order to fail faster – in the name of progress and productive investments that help the company and end consumers.
The FDA’s ICH Q10 “Process Performance and Product Quality Monitoring System” section says, “Pharmaceutical companies should plan and execute a system for the monitoring of process performance and product quality to ensure a state of control is maintained. An effective monitoring system provides assurance of the continued capability of processes and controls to produce a product of desired quality and to identify areas for continual improvement.” (3)
Added Value Beyond Compliance
Meeting compliance requirements historically required mandatory investments in technology systems. Progressive companies who have extended the usefulness of their systems to add business value – by making their entire operations more efficient and compliant, for example – have realized benefits across their organizations. Taking a risk-based approach to manufacturing and distribution during the entire lifecycle of a product helps their scientists and other individuals involved in product lifecycle development.
Tools, such as electronic lab notebooks, bring value by sharing data from past experiments with research colleagues. If records were stored on only one laptop – or even buried in a server folder – valuable notes might be lost and experiments unnecessarily repeated. Data trending through analysis with the right software tools can provide key information to allow risk mitigation, risk-based management, and continuous process improvements. Whether through quality management, process and adverse event reporting, enterprise resource management, and/or Laboratory Information Management Systems (LIMS) implementation, companies can view their drug development processes holistically using real-world input.
Collecting, storing, and sharing data from early drug development research and process design through manufacturing quality and process improvement requires a risk-based approach. This approach must control, contain, and ensure compliance during the entire product lifecycle. In addition, this must cross all facilities in a closed loop that can lead to failing faster when that is the inevitable outcome, and, in turn, can get to the end goal faster: more efficient drug release and production.
REFERENCES
- Medicine.Net, “Drug Approvals - From Invention to Market ... A 12- Year Trip,” http://www.medicinenet.com/script/main/art.asp?articlekey=9877
- FDA Review, The Independent Institute, “The Drug Development and Approval Process” http://www.fdareview.org/approval_process.shtml
- “Guidance for Industry Q10 Pharmaceutical Quality System,” U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), April 2009