
Best Practices For Ensuring Quality In Biologic Products

Given the growing complexity of new biologic therapies and the growing ubiquity of first generation biologics, new challenges continue to emerge and drug manufacturers and compounders alike need to think differently about the packaging and delivery of these therapies. Because these biologic entities are complex, their stability risks are higher and because they are large in size, their immunogenicity risk is higher when compared to small molecules. Therefore, the objective of manufacturers when it comes to packaging and delivery of biologics is to reduce risk to the fullest possible extent. Whether the challenge is to provide robust cryopreservation containment for gene or cell therapies, to develop wearable injectors to transition oncology mAbs out of infusion clinics and into a home setting, or to modernize the packaging systems used by 503B outsourcing facilities, there are several universal best practices for ensuring the quality of the biologic product.
Material Selection. Selecting appropriate materials for packaging biologics is the most basic consideration in reducing stability risks. Biologic drugs are complex entities which are susceptible to instabilities when exposed to light, elevated temperatures or temperature changes, leachables, reactive gases, formulation changes over time, and other external stressors. For this reason, most biologic drug manufacturers typically select materials which present the lowest possible risk of interaction such as barrier coated elastomeric closures and low surface energy container materials, while simultaneously seeking to minimize or eliminate silicone oil in these systems.
Especially in the compounding industry, the use of containers that are not intended for long term contact (such as heavily siliconized polypropylene syringes) presents the risk of unforeseen stability issues and potentially shorter Beyond Use Dates (BUD's). The FDA's guidance, "Container Closure Systems for Packaging Human Drugs and Biologics" states that each Biologics License Application (BLA) should include an analysis of the suitability of the container systems, the stability (of the drug in the container system and of the container system itself) and a quality control strategy. While less
stringent than the requirements for a BLA, the requirements for compounders (operating outside the scope of a BLA), are beginning to align more closely. In January 2018, the FDA issued a final guidance on "Mixing, Diluting, or Repackaging Biological Products Outside the Scope of an Approved Biologics License Application." A noteworthy part of this guidance
is Appendix A which describes the establishment of a Beyond Use Date for the repackaged biologic drug based on stability testing. This provides formal guidance for stability testing that is reminiscent of the expectations of a BLA. As examples, in this document, subvisible particle levels must be tested and USP<788> and USP<789> requirements are referenced. USP <1207> is referenced for establishing Container Closure Integrity (CCI) and the maintenance of sterility. Also, product potency and product related impurities (including protein aggregates, charge variants, etc.) must be characterized in order to assign a Beyond Use Date. Compounders may be able to reduce stability risks and possibly extend BUD's by moving away from low quality disposable packaging in favor of traditional parenteral packaging options. (This statement is likely true not only for biologics but for small molecule compounded drugs as well.)
Figure 1 shows several example impacts that material selection can have on attributes such as extractables and protein aggregation. In Fig. 1a, gas chromatograms of an n-heptane extraction solvent that has come into contact with an uncoated elastomeric closure (top) versus a fluoropolymer film coated elastomer (bottom) show numerous organic extractables originating from the rubber which can be blocked by the presence of the barrier film. Some biologics, such as Orencia® as seen in Fig. 1b, are prone to aggregation in the presence of silicone oil (the gray bars show Orencia® aggregation in siliconized glass syringes). By eliminating silicone oil altogether, the aggregation can be mitigated (as shown by the blue bars indicating unsiliconized cyclic olefin polymer (COP) syringes). Finally, there is growing appreciation that the surface energy of the container can impact protein aggregation. In Fig. 1c, vials made of glass (which has a highly negatively charged surface) are associated with higher degrees of aggregation of the selected model protein (with agitation and in the absence of surfactant) as compared to COP vials (which have a low surface energy.). Selecting primary packaging materials with low surface energy (such as COP) and those materials which eliminate silicone oil may help drug manufacturers and compounders alike to meet USP<788> and USP<789> particle level requirements.
System Integration. Another best practice to ensure quality and reduce risks in biologic drug development is to consider, at the earliest possible stages, the packaging and delivery from a full integrated systems perspective. From the simplest container systems such as vials to the most complex delivery systems such as wearable injectors, all pieces of the packaging / delivery device should be designed and should operate as an integral system: the components must work in concert with the drug and with each other, under applicable storage and delivery conditions. The supplier of the packaging system plays a key role in this integrated system approach.
Figure 2 shows several examples of when full system integration needs to be considered in biologics packaging. Most gene and cell therapies will initially go to market packaged in vials which are cryogenically stored at either -80ºC or -180ºC until delivery to the patient. Under these harsh storage conditions, polymer materials such as halobutyl elastomers used in vial stoppers will experience volume shrinkage in accordance with the material's coefficient of thermal expansion (CTE). As seen in Fig. 4a, the volume shrinkage is similar for rubber stoppers and for cyclic olefin polymer vials (approximately 4.5% and 4.1% respectively in transition from room temperature to -180ºC.). Glass however shows little volume shrinkage. The mismatch in CTE's for rubber and glass presents a higher risk for container closure integrity breaches during cooling, storage, and warming as compared to the combination of rubber and COP. This has previously been observed experimentally through oxygen headspace analysis and is summarized in Fig. 2a. Even in the simplest case of vial containment, it is critical to understand how the components of a packaging system work together and it is helpful if a packaging supplier can provide data packages to evidence the performance of the integrated system.
As another example in Fig. 2b, a drug manufacturer filing a combination product that uses an autoinjector to deliver a viscous drug must understand how the drug viscosity impacts the syringe delivery forces and therefore the autoinjector design requirements. If such data (using a placebo solution for example) can be provided by the syringe manufacturer, it can help to de-risk the combination product development, reduce the time to market, and help to ensure the most reliable device performance. Wearable injectors can deliver large drug volumes over an extended period of time and may offer a solution for moving oncology biologics out of the clinic and into a homecare setting. Reliable performance of such a complex device (for example, consistent injection time as in Fig 4c), is the culmination of successful system integration of the drug, primary packaging, and delivery device.
With the growing complexity of new therapeutic biologic entities and the growing ubiquity of existing entities, it is becoming more important than ever for drug manufacturers and compounders alike to reduce risks. Choosing the appropriate packaging materials and considering full system integration at the earliest stages are two foundational best practices for ensuring quality. While accountability for the safety, compatibility and performance of a combination product lies solely with the drug manufacturer who is filing the drug with the regulators, or with the compounder who is repackaging the drug, responsibility for the full system characterization is becoming more broadly distributed across the supply chain. The supplier of the container system and or device should understand safety, compatibility and performance from a full system perspective and can help to supply the documentation or analytical services required to support the combination product development.
Acknowledgements: Many thanks to Cathy Zhao, Lloyd Waxman, Ranjana Singh, Le Ho, Page McAndrew, and the entire West Analytical Laboratory Services team for their contributions to this work.
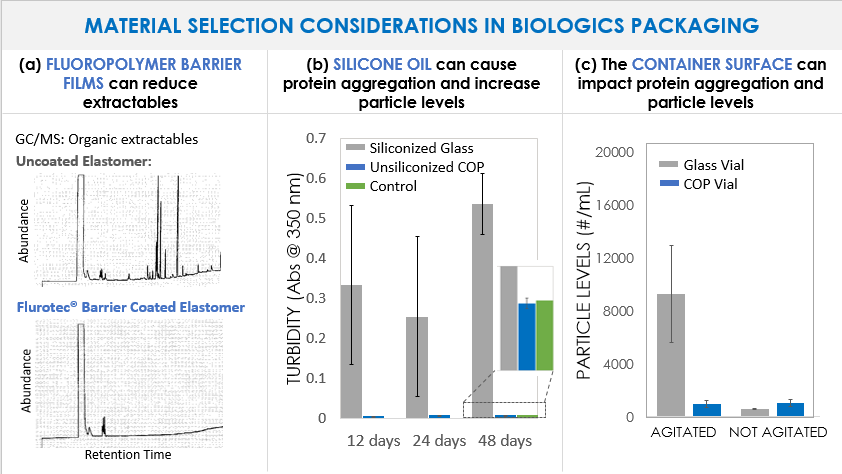
Figure 1. Examples of the impacts that primary packaging material selection can have on biologic drug formulations. (a) Gas chromatograms of an n-heptane extraction solvent after exposure to uncoated (top) and Daikyo Flurotec laminated (bottom) elastomeric closures. (b) Turbidity measured at by absorbance at 350nm of Orencia® formulations in siliconized glass syringes (gray bars) and unsiliconized Daikyo Crystal Zenith® cyclic olefin polymer (COP) syringes (blue bars). Syringes have been agitated by end over end rotation for up to 48 days. The control (green bar) represents the unagitated formulation stored in a glass vial in the refrigerator. (c) Particle levels (#/mL) measured by flow imaging in a Humanized IgG1 antibody formulation (143 kDa, pI = 8.4, pH = 6.9, no surfactant) stored in glass vials (gray bars) or Daikyo Crystal Zenith® cyclic olefin polymer vials (blue bars). The left two bars have been agitated on an orbital shaker table at 200 rpm for 4 days at room temperature while the right two bars are unagitated.
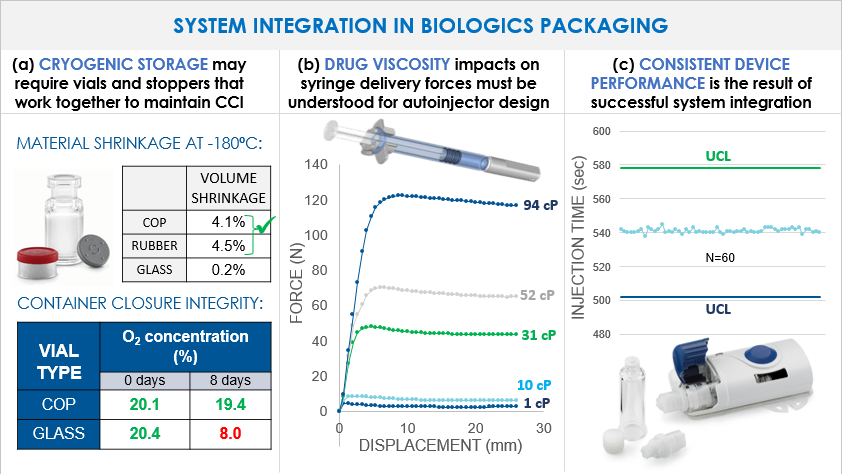
Figure 2. (a) Top - Calculated % volume shrinkage of COP, rubber, and glass (coefficients of thermal expansion of 70, 77, and 4 * 10-6cm/cm-K respectively) upon freezing from room temperature to -180ºC. Bottom – contain closure integrity (CCI) testing by oxygen headspace analysis at time 0 and 8 days cryogenic storage of a COP vial versus a glass vial with the same elastomeric closures. The glass vial shows a decrease in % oxygen indicating a CCI breach. (b) Delivery force profiles (200 mm/min) in a 1 mL long Daikyo Crystal Zenith® syringe with a 27G thin walled needle at varying solution viscosities from 1 to 94 cP, aged for 3 months at room temperature. (2) Injection times of 60 different SmartDose™ wearable injector devices as compared to the upper and lower control limits. Consistency in injection time is ensured in part by displacement-controlled (rather than force-controlled) administration and demonstrates successful device integration.

- https://www.fda.gov/Drugs/DrugSafety/ucm458952.htm
- https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070551.pdf
- https://www.fda.gov/downloads/drugs/guidances/ucm434176.pdf
- Orencia® is a trademark of Bristol-Meyers Squibb Company.
- W. Winters. When Glass Vials Fail at Low Temperatures, Consider A Cyclic Olefin Polymer System. https://www.pharmaceuticalonline.com/doc/when-glass-vials-fail-at-low-temperatures-consider-a-cyclic-olefin-polymer-system-0001
- Daikyo Flurotec® is a registered trademark of Daikyo Seiko, Ltd. in Japan.
- Crystal Zenith® is a registered trademark of Daikyo Seiko, Ltd.
- SmartDose is a trademark of West Pharmaceutical Services, Inc. in the United States and other countries.