By Kelly Jordan, Principal CSV Consultant, ProPharma Group
Data integrity is a current hot topic, but not a new one, within the life sciences industries and associated product supply chains.
This article will not delve into why there have been so many recent data integrity issues within the EU and FDA regulated industries. Instead, this article will aim to gist the regulatory perspective and identify current best practice thinking relative to what one can do from a compliance and quality perspective to avoid and detect data integrity issues and overall data quality pitfalls.
If you have read regulatory guides and rules, you have read the generic and recurring terms…’quality and integrity of the data’…
So what does that mean?
Just as pharmaceutical products must meet certain quality attributes associated with effects on patients such as strength, identity, safety, purity, and quality (SISPQ), so too must the associated data meet certain quality and integrity attributes…i.e., ALCOA+.
The acronym ALCOA6 stands for the following attributes: Attributable, Legible, Contemporaneous, Original, and Accurate. Refer to the glossary at the end for definitions of terms.
ALCOA may be considered the data quality attributes focused on doing it right the first time when it is done, i.e., task based.
The acronym ALCOA+7 stands for ALCOA in addition to the following attributes: Complete, Consistent, Enduring, and Available. Again, refer to the glossary at the end for definitions of terms.
ALCOA+ may be considered the data quality attributes that are focused on establishing and monitoring the support processes around data activities, continuous improvement and overall product quality.
So in order to achieve overall data quality and associated product quality, one must have both ALCOA and ALCOA+. Therefore, one can infer that Product Quality is directly associated with Data Quality.
This association of ALCOA, ALCOA+ and overall data and product quality can be theoretically depicted by the diagram below. It shows that the higher the ALCOA and ALCOA+, the higher the overall data quality and product quality.
![]()
![]() In order to further set the regulatory context, the following excerpts from the FDA Guidance for Industry – Electronic Source Data in Clinical Investigations provide a mindset that can be transferrable to any computer system intended to satisfy data integrity expectations:
In order to further set the regulatory context, the following excerpts from the FDA Guidance for Industry – Electronic Source Data in Clinical Investigations provide a mindset that can be transferrable to any computer system intended to satisfy data integrity expectations:
…capturing source data in electronic form…is intended to assist in ensuring the reliability, quality, integrity, and traceability of data from electronic source to electronic regulatory submission.
… Adequate controls should be in place to ensure confidence in the reliability, quality, and integrity of the electronic source data.
Assuring data integrity requires appropriate quality and risk management systems and processes, including adherence to sound scientific principles and good (electronic) documentation practices, keeping in mind that there should be no loss of quality when an electronic system is used in place of a paper system.
Furthermore, technology alone cannot eliminate data integrity issues. There are still people and manual processes involved that must be accounted for, monitored, and improved.
![]() Therefore, if people and manual processes are still involved to some degree, one must take a holistic approach to addressing data integrity and applying the necessary designs and controls across all the spheres of influence depicted below.
Therefore, if people and manual processes are still involved to some degree, one must take a holistic approach to addressing data integrity and applying the necessary designs and controls across all the spheres of influence depicted below.
![]() So where and how should one implement data integrity and data quality design considerations and controls?
So where and how should one implement data integrity and data quality design considerations and controls?
The table below identifies areas of focus and consideration.
One important caveat, when trying to implement a system and associated controls, is that a team is involved throughout the entire lifecycle. It must be a team of qualified subject matter experts (SMEs) in the following minimum areas: System/Business Process Owner, Technical Representative, Computer System Validation, and Quality.
|
Example Data Quality Concern
|
Design/Implementation Considerations
|
Verification Considerations
|
Pitfalls to Avoid / Management Considerations
|
|
Attributable - who acquired the data or performed an action (or modification) and when
|
Given the inherent increase in complexity from a paper system, computerized systems must be defined, procured, and configured to meet the applicable regulatory record requirements.
Applicable Tasks/Deliverables:
- System requirements/user requirements and/or assessments must define the intended system record types and Annex 11/21 CFR Part 11 applicability.
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Design/configuration documentation should include the specific post-installation attributes that must be set to meet the requirements of the system.
|
- There should be definition of separate user roles (based on record involvement) for all systems (e.g., Creator/User ability to write records, Reviewer ability to append/modify a record, and Administrator ability to delete).
- Each user regardless of role must have a unique user ID to access the system. Typically this is handled most efficiently via a centralized network (e.g., Active Directory Groups).
- An Annex 11/Part 11 assessment should be performed to verify all expected regulatory requirements including audit trail and electronic record/signature attributes where applicable (e.g., secure).
- Security settings within the application must be designed and configured to not allow non-administrators the ability to disable compliance related settings such as those related to audit trail, user management, and signatures.
- Security settings outside of the application must be designed and configured to only allow the minimum user permissions for the application to function.
Specific considerations:
- If a database is used, have it located and administered on a qualified infrastructure independent from the application client computer with no user access to the raw data/database location outside of the application.
- If the computer system is standalone, security controls need to be in place to limit user ability to modify or delete raw data, metadata, and audit trail information.
|
- Users of the system should not have more than one role.
- If possible utilize a system administrator who is independent from the department responsible for electronic records (e.g., IT) or one that does not have a vested interest in the data results from the given system.
- System administrators should not generate or review data.
- Users who are not Administrators should not be in a Local Administrators group or Power/Super Users group.
- The use of shared and generic log-on credentials must be avoided to ensure that personnel actions documented in electronic records and signatures can be attributed to a unique individual.
- Implement SOP direction (and associated training) to identify the importance of data integrity and define procedural controls as necessary to secure the data flow within the process and prohibit the overwriting or deleting of data. For some (e.g., standalone) systems, this may involve a hybrid electronic/paper approach and maintaining a continuous session on the system with additional reviews if needed.
|
|
Legible - Data is permanent and easily read (by a human)
|
Applicable Tasks/Deliverables:
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Design/configuration documentation should include the specific post-installation attributes that must be set to meet the requirements of the system.
|
- Controlled configuration and use of any record annotation tools in a manner that prevents data in display and print from being obscured (if possible).
- Verification of record/report output against on-screen or originally entered (meta) data.
|
- Implement SOP direction (and associated training) to identify the importance of data integrity and define procedural controls as necessary to direct that no data is to be obscured during use and output (e.g., hardcopy) is reviewed where applicable for legibility and consistency with good documentation practices.
|
|
Contemporaneous - documented at the time of the activity (promptly)
|
Applicable Tasks/Deliverables:
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Design/configuration documentation should include the specific post-installation attributes that must be set to meet the requirements of the system.
- Specific consideration needs to be made relative to what centralized (e.g., domain) security can be implemented versus standalone/local security configuration.
- Specific consideration needs to be made relative to the need for and definition of centralized and synchronized time stamping.
|
- An Annex 11/Part 11 assessment should be performed to verify all expected regulatory requirements including audit trail and electronic record/signature attributes where applicable (e.g., secure).
- Verification needs to be made that users cannot change system date, time and time zone on the computer that the application uses to stamp that information.
- This can be controlled centrally for example by defining a specific organizational unit (group) where computers in this group only allow a network administrator to change these attributes.
- If this has been handled locally on the standalone computer, users cannot be in the local administrators group.
- If the system is an enterprise level system where use may span multiple time zones, verification needs to be made relative to a consistent centralized time for the system regardless of access point and that time is synchronized to a traceable source.
|
- Users who are not Administrators should not be in a Local Administrators group or Power/Super Users group.
- Implement SOP direction (and associated training) to identify the importance of data integrity and overall good documentation practices (even if the computer system is providing the necessary information).
|
|
Original - the first recording of data, raw or source data, or a certified true copy
|
Applicable Tasks/Deliverables:
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Design/configuration documentation should include the specific post-installation attributes that must be set to meet the requirements of the system.
- Specific consideration needs to be made relative to what centralized (e.g., domain) security can be implemented versus standalone/local security configurations.
- If a standalone system, consideration needs to be made relative to process/procedural data flow and if automatic or manual functions will be needed. Folders may need to be set for either write permissions with deny append or read and execute with allow to write. Also permission inheritance needs to be accounted for.
|
- An Annex 11/Part 11 assessment should be performed to verify all expected regulatory requirements including audit trail and electronic record/signature attributes where applicable (e.g., secure).
- Security settings outside of the application must be designed and configured to only allow the minimum user permissions for the application to function.
Specific considerations:
- If a database is used, have it located and administered on a qualified infrastructure independent from the application client computer with no user access to the raw data/database location outside of the application.
- In a standalone situation, a user should not have full control of the records or audit trail location. Ideally users should only have read, write, execute permissions with the functional denial of modify/write-over and delete.
|
- A formal system development/implementation life cycle should be followed and if process specific considerations/evaluations are needed, a development or test environment is advantageous for proving out a design prior to formal end-use environment verification.
- A procedure or procedures (and associated training) should dictate the acceptable and consistent data management practices for the system including how an original data record is processed/saved, how it may be reviewed or how it can have metadata associated with it (e.g., signed), and how it can be historically retrieved, backed-up, and restored.
|
|
Accurate - data is correct including context/meaning (e.g., metadata) and edits
|
Applicable Tasks/Deliverables:
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Design/configuration documentation should include the specific post-installation attributes that must be set to meet the requirements of the system.
- Specific consideration needs to be made relative to defining any custom or process-specific calculations, reporting, or critical process parameters/critical quality attributes that may require data validation, calibration, or supplemental risk assessment and verification.
|
- An Annex 11/Part 11 assessment should be performed to verify all expected regulatory requirements including audit trail and electronic record/signature attributes where applicable (e.g., secure).
- Specific verification/validation documentation (e.g., test plans, scripts, protocols, traceability matrix) should define, challenge, confirm, and trace the accuracy of the defined data collection, processing, and reporting for the system.
|
- A procedure or procedures (and associated training) should dictate the acceptable and consistent data management practices for the system including how an original data record is processed/saved, how it may be reviewed or how it can have metadata associated with it (e.g., signed), and how it can be historically retrieved, backed-up, and restored.
- A procedure or procedures (and associated training) should dictate the required calibration frequency of any instrumentation associated with critical data in the system.
|
|
Complete - Data includes all data (passing or otherwise) from all actions taken to obtain the required information, including metadata (e.g., audit trail) and edits
|
Applicable Tasks/Deliverables:
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Design/configuration documentation should include the specific post-installation attributes that must be set to meet the requirements of the system.
|
- An Annex 11/Part 11 assessment should be performed to verify all expected regulatory requirements including audit trail and electronic record/signature attributes where applicable (e.g., secure).
|
- A procedure or procedures (and associated training) should clearly identify the company’s data integrity definitions and expectations.
- A procedure or procedures (and associated training) should clearly dictate the acceptable and consistent data management practices for the system including how an original data record is processed/saved, how it may be reviewed or how it can have metadata associated with it (e.g., signed), and how it can be historically retrieved, backed-up, and restored.
- A procedure or procedures (and associated training) should dictate the required steps for addressing out of tolerance results and process deviations.
- A procedure or procedures (and associated training) should dictate the required steps for how to perform an audit trail review and at what frequency.
|
|
Consistent - Data is created in a repeatable and comparative manner (traceable)
|
Applicable Tasks/Deliverables:
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Design/configuration documentation should include the specific post-installation attributes that must be set to meet the requirements of the system.
- Specific consideration needs to be made relative to defining any custom or process-specific automated processing/workflows or specific sequencing of events.
|
- Specific verification/validation documentation (e.g., test plans, scripts, protocols, traceability matrix) should define, challenge, confirm, and trace the consistency of the defined data collection, processing, and reporting for the system.
- This may include but not be limited to the following:
- Equipment/Instrument qualification
- Software/System qualification
- Method Validation
- Process Validation
|
- Formal policy, plan or procedure documentation should exist (along with associated training) dictating the system development and maintenance life cycle along with the applicable method and process validation expectations associated with the system.
- A procedure or procedures (and associated training) should clearly dictate the acceptable and consistent data management practices for the system including how an original data record is processed/saved, how it may be reviewed or how it can have metadata associated with it (e.g., signed), and how it can be historically retrieved, backed-up, and restored.
|
|
Enduring - Stored on media proven for the record retention period
|
Applicable Tasks/Deliverables:
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Requirements/design/configuration documentation should include the specific post-installation attributes that must be set to meet the intended use/future state functions of the system.
- Specific consideration and definition needs to be made relative to identifying the specific record types (e.g., records dictated by regulation, i.e., predicate rule) and what is the record’s respective retention period.
- Specific consideration and definition needs to be made relative to what technology and media type will best satisfy the day-to-day use and long-term retention/usability of the affected stored records.
|
- Specific verification/validation documentation (e.g., test plans, scripts, protocols, traceability matrix) should define, challenge, confirm, and trace the ability of the system to store and retrieve records for the entire duration of a record’s retention period.
- This may include but not be limited to the following:
- Vendor/Media life span information
- Media reliability information
- Media use schedules
- Restoration verification
|
- Formal policy, plan or procedure documentation should exist (along with associated training) dictating the minimum retention period for the record types affected by the system.
- A procedure or procedures (and associated training) should clearly dictate the acceptable and consistent data backup processes, schedules, media types, on-site and off-site schedules, archive, and restoration activities.
|
|
Available - Readily accessible in human readable form for review throughout the retention period for the record
|
Applicable Tasks/Deliverables:
- Prior to procurement, a vendor assessment and a product demonstration should be performed to aid in determining design/configuration needs and any potential compliance risks.
- Requirements/design/configuration documentation should include the specific post-installation attributes that must be set to meet the intended use/future state functions of the system.
- Specific consideration and definition needs to be made relative to identifying the specific record types (e.g., records dictated by regulation, i.e., predicate rule) and what is the record’s respective retention period.
- Specific consideration and definition needs to be made relative to what technology and media type will best satisfy the day-to-day use, long-term retention capability, and retrieval time requirements.
|
- Specific verification/validation documentation (e.g., test plans, scripts, protocols, traceability matrix) should define, challenge, confirm, and trace the ability of the system to store records for the entire duration of a record’s retention period and be able to retrieve/restore in the time frame necessary for internal and external review.
- This may include but not be limited to the following:
- Verification of restoration capability from all media types
- Verification of restoration capability from all backup schedules (e.g., on-site and off-site)
- Verification of archive retrieval capability
|
- Formal policy, plan or procedure documentation should exist (along with associated training) dictating the minimum retention period for the record types affected by the system.
- A procedure or procedures (and associated training) should clearly dictate the acceptable and consistent data backup processes, schedules, media types, on-site and off-site schedules, archive, and restoration activities.
- Procedural and periodic tests should be performed to verify the ability to retrieve archived electronic data from storage locations.
- Considerations for decommissioned/retired or archived systems, there may be need for provisioning of suitable reader equipment, such as software, operating systems and virtualized environments, etc., to view the archived electronic data when required.
|
In summary, data integrity is a component of data quality that is directly relational to product quality. Technology alone will not solve the situation; it requires a hybrid (human and computerized) approach to address and improve your overall data and product quality. That said, if one considers and implements systems in a team-based approach, focused on process/product knowledge and continuous improvement, positive results will be seen relative to increased efficiency, compliance, and quality.
Glossary:
Accurate: data is correct including context/meaning (e.g., metadata) and edits
ALCOA: Attributable, Legible, Contemporaneous, Original, and Accurate
ALCOA+: ALCOA in addition to the following attributes: Complete, Consistent, Enduring, and Available
Attributable: who acquired the data or performed an action (or modification) and when
Available: Readily accessible in human readable form for review throughout the retention period for the record
Complete: Data includes all data (passing or otherwise) from all actions taken to obtain the required information, including metadata (e.g., audit trail) and edits
Consistent: Data is created in a repeatable and comparative manner (traceable)
Contemporaneous: documented at the time of the activity (promptly)
Enduring: Stored on media proven for the record retention period
Integrity: The extent to which all data are complete, consistent and accurate throughout the data lifecycle. MHRA and WHO definitions
Legible: Data is permanent and easily read (by a human)
(data) Lifecycle: A planned approach to assessing and managing risks to data in a manner commensurate with potential impact on patient safety, product quality and/or the reliability of the decisions made throughout all phases of the process by which data is created, processed, reviewed, analyzed and reported, transferred, stored and retrieved, and continuously monitored until retired MHRA / WHO definition
Original: the first recording of data, raw or source data, or a certified true copy
(data) Quality: [ICH Q10] The degree to which a set of inherent properties of a product, system or process fulfils requirements.
Source Data (clinical trial): All information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical trial necessary for the reconstruction and evaluation of the trial. [FDA][
About Author:
Kelly Jordan is a Principal CSV Consultant for ProPharma Group. He holds an undergraduate degree in Electrical and Electronic Engineering Technology. Kelly has over 20 years of computer system validation experience in biopharmaceutical, pharmaceutical, and medical device environments. His experience includes individual contributor work on all GAMP categories of computerized systems from instruments and infrastructure to enterprise level applications and information systems. Kelly has also lead, managed, and participated in teams that have implemented and operated quality management systems for compliance services, IT infrastructure, and software development. Kelly is a past senior member of PDA. He is a current member of ASTM with multiple committee membership and is a member of ISPE, where he participates in multiple communities of practice.
References:
- FDA, Guidance for Industry – Computerized Systems Used in Clinical Investigations, May 2007
- FDA, Guidance for Industry – Electronic Source Data in Clinical Investigations, September 2013
- MHRA, MHRA GMP Data Integrity Definitions and Guidance for Industry, March 2015
- Newton, M., White, C. “Data Quality and Data Integrity: What is the Difference?”, ispeak, June 2015
- WHO, “Guidance on Good Data and Record Management Practices” September 2015
- Woollen, Stan W. “Data Quality and the Origin of ALCOA” The Compass – Summer 2010
- White, Christopher H., Gonzalez, Lizzandra R. “The Data Quality Equation – A Pragmatic Approach to Data Integrity | IVT, August 2015.


 In order to further set the regulatory context, the following excerpts from the FDA Guidance for Industry – Electronic Source Data in Clinical Investigations provide a mindset that can be transferrable to any computer system intended to satisfy data integrity expectations:
In order to further set the regulatory context, the following excerpts from the FDA Guidance for Industry – Electronic Source Data in Clinical Investigations provide a mindset that can be transferrable to any computer system intended to satisfy data integrity expectations: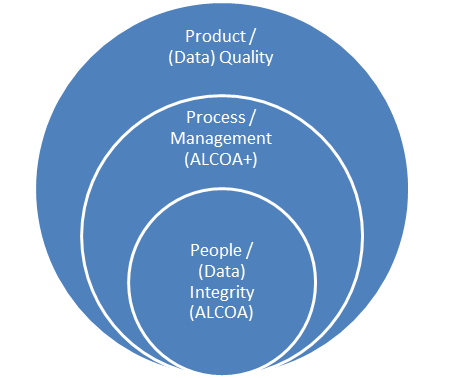 Therefore, if people and manual processes are still involved to some degree, one must take a holistic approach to addressing data integrity and applying the necessary designs and controls across all the spheres of influence depicted below.
Therefore, if people and manual processes are still involved to some degree, one must take a holistic approach to addressing data integrity and applying the necessary designs and controls across all the spheres of influence depicted below. So where and how should one implement data integrity and data quality design considerations and controls?
So where and how should one implement data integrity and data quality design considerations and controls?