
Building Quality Into Radiopharmaceuticals: A Look At EMA's Recent Guideline
By Tim Sandle, Ph.D.

The draft Guideline on quality of radiopharmaceuticals1 issued by the European Medicines Agency (EMA) and adopted by the Committee for Medicinal Products for Human Use (CHMP) provides an update to the European regulatory expectations for the quality documentation of radiopharmaceuticals. The draft guideline, released for public consultation in December 2025, replaces the earlier 2007 guideline and reflects significant scientific, technological, and regulatory developments in the field of radiopharmaceuticals over the last two decades.
Radiopharmaceuticals are recognized as a distinct class of medicinal products under Directive 2001/83/EC.2 Their unique characteristics include the presence of radionuclides, strength defined in terms of radioactivity rather than mass, rapid radioactive decay, and — in many cases — very short shelf lives. These features necessitate specific adaptations to conventional pharmaceutical quality principles, particularly concerning manufacture, control, stability, and release.
The guideline focuses on the quality part of the ICH Common Technical Document (CTD),3 primarily Module 3, and explains how established EU and ICH pharmaceutical guidelines should be interpreted or adapted for radiopharmaceuticals. It applies both to marketing authorization applications and to variations for authorized products.
Scope Of The Guideline
The scope of the guideline covers four main categories of radiopharmaceutical-related products:
- Ready‑for‑use radiopharmaceuticals, including positron emission tomography (PET) radiopharmaceuticals
- Kits for radiopharmaceutical preparation, which are radiolabeled at the point of use
- Radionuclide generators, from which a daughter radionuclide is eluted
- Radionuclide precursors, intended for radiolabeling other substances
In addition, the guideline applies to substances used in the manufacture of radiopharmaceuticals, including active substances in kits, chemical precursors, and radionuclide precursors used as starting materials. However, radiopharmaceuticals based on monoclonal antibodies are explicitly excluded since they are covered by a separate guideline.
The principles described are also applicable — using a phase‑appropriate and flexible approach — to radiopharmaceuticals used in clinical trials.
Key Concepts And Definitions
The guideline emphasizes several fundamental principles that distinguish radiopharmaceuticals from conventional medicinal products. These are:
- Radioactivity expression must be in becquerels (Bq) (the activity of a quantity of radioactive material in which one nucleus decays per second)4 and always linked to a defined activity reference date and time, including time zone where applicable.
- Posology (dosage) is based on administered radioactivity, not mass or molar amount. This is because the therapeutic or diagnostic effect of the product depends on the energy deposited by the radiation, not the quantity of molecules present.5
- Radioactive decay, which means product quality, strength, and impurities evolve continuously after manufacture.
- Radiolabeled active substances are often not isolated. The draft text indicates that quality assurance must therefore be achieved via upstream control of precursors and processes.
The text also clarifies that most diagnostic radiopharmaceuticals have no direct pharmacodynamic effect; therapeutic radiopharmaceuticals rely on radiation as the primary mode of action.
Active Substance: General Regulatory Approach
With conventional medicines, the active substance is administered to the patient. However, for radiopharmaceuticals, this paradigm is adapted. To demonstrate this, the guideline differentiates between:
- the radiolabeled active substance, which is often transient and not isolable, and
- chemical precursors and radionuclide precursors, which are isolable, testable, and form the basis for quality assurance.
From a regulatory perspective, these substances are treated as active substances or active substance intermediates. Hence, their manufacturers must comply with GMP for active substances and be listed in the dossier.
Radionuclide Precursors
Radionuclide precursors are the foundational components in nuclear medicine. These are defined as radioactive isotopes produced specifically for the radiolabeling of another substance (vector) before administration. These are typical solutions containing a radionuclide in a chemical form.6 They may be:
- used directly in ready‑for‑use radiopharmaceutical manufacture
- marketed as radionuclide precursors
- incorporated into radionuclide generators.
The guidelines describe the key requirements for radionuclide precursors, including:
- full description of origin (such as cyclotron, reactor, or mass separation)
- decay characteristics (such as half‑life, decay chain, and radiation types)
- detailed manufacturing processes, including irradiation parameters, target materials, separation chemistry, and automated synthesis modules, where used
- control of radionuclidic, radiochemical, and chemical impurities
- justification of container closure systems for radioactive solutions
- stability data is appropriate to radioactive materials (this section acknowledges that traditional stability paradigms do not fully apply).
Chemical Precursors And Kit Active Substances
For kits, the active substance is the nonradioactive component designed to bind the radionuclide. For ready‑for‑use radiopharmaceuticals, chemical precursors represent the closest nonradioactive analogues of the radiolabeled active substance.7
In relation to this, the guideline requires:
- selection of structurally relevant precursors
- compliance with general active substance quality guidelines, where applicable
- control of relevant impurities based on safety, radiochemical purity, and impact on biodistribution
- application of European Pharmacopoeia monographs where available.
Radiolabeled Active Substances
Where the radiolabeled active substance cannot be isolated, a reduced pathway can be provided as part of the regulator submission. This must still include:
- identification of radionuclide position
- source and decay properties
- specification parameters, even if tested indirectly
- justification of quality assurance through process development, validation, and in‑process controls.
Drug Product: Ready‑For‑Use Radiopharmaceuticals And Radionuclide Precursors
For ready‑for‑use products, the drug product is often manufactured in a continuous process, particularly for PET radiopharmaceuticals. To assist manufacturers, the guideline focuses on:
- clear description of composition, with radioactivity and mass both stated
- justification of proposed strength(s) or strength ranges, considering short half‑lives and ease of administration
- consideration of radiolysis and the use of stabilizers or radical scavengers
- validation of compatibility with containers, closures, syringes, and shielding
- strict control of raw materials and disposables, particularly when products are released before all test results are available.
For sterile filtered products, bioburden controls must ensure ≤10 CFU/100 mL prior to sterilizing filtration. This matches the standard expectations for aseptically filled products.
Control Of The Drug Product
In developing control attributes, the guideline indicates what is expected of the product specifications. This must include, as appropriate:
- radiochemical identity and purity
- radionuclidic identity and purity
- radioactive concentration
- chemical purity
- sterility and bacterial endotoxins for parenteral products.
For sterile products, the guideline recognizes that the sterility test may be performed post‑release. This is due to the short shelf life and the patient need. While this option is provided, the guidance emphasizes the need for validated processes (which should connect into a contamination control strategy) together with a robust retrospective review led by quality.
Acceptance limits are specified more narrowly than for conventional medicines:
- Diagnostic radiopharmaceuticals: 90%–110% of the label claim
- Therapeutic radiopharmaceuticals: 95%–105% of the label claim
The annex to the guidance document provides a consolidated list of minimum specification parameters applicable to each category:
- ready‑for‑use radiopharmaceuticals
- radionuclide precursors
- radionuclide generators
- kits for radiopharmaceutical preparation.
The parameters described include radionuclidic identity and purity, radiochemical identity and purity, chemical purity, radioactivity, sterility, endotoxins, particulate matter, and excipient assays where relevant. The annex clarifies where testing may be performed upstream rather than on the final product, provided this is scientifically justified.
Stability
Stability requirements are needed, as with any conventional pharmaceutical. However, these are adapted in the guidance to account for radioactive decay. The core requirements are:
- Shelf‑life is usually defined from the end of manufacture or activity reference time.
- Full ICH stability study durations do not apply to products with noticeably short shelf lives.
- Testing frequency and duration must be justified based on real‑world use.
- For multidose preparations, in‑use stability must be evaluated, typically with an upper limit of 8 hours unless justified.
Radionuclide Generators
Radionuclide generators are portable, shielded devices used in nuclear medicine to produce short-lived radioactive isotopes (daughters) from longer-lived parent radionuclides on-site.8 These are treated in the guideline as complex systems comprising:
- the parent radionuclide
- the daughter radionuclide eluate
- the eluent and associated accessories.
To manage these, the guideline requires:
- detailed description of generator design, column materials, and stationary phase
- control of parent radionuclide breakthrough
- validation of sterility, eluate purity, and elution performance over shelf life
- stability data addressing aging and the effect of repeated elutions
- clear instructions and illustrations in the summary of product characteristics (SmPC) to minimize misuse and contamination.
Kits For Radiopharmaceutical Preparation
For kits, the guideline sets out stringent expectations to ensure reproducibility at the user level. These expectations comprise:
- clear specifications of radionuclide precursors or generators suitable for labeling
- validation of labeling procedures under worst‑case conditions (activity, volume, shelf life)
- defined end user quality control tests with validated acceptance criteria
- stability data for both the marketed kit and the radiolabeled preparation, including in‑use stability
- explicit SmPC statements covering shelf life before and after radiolabeling.
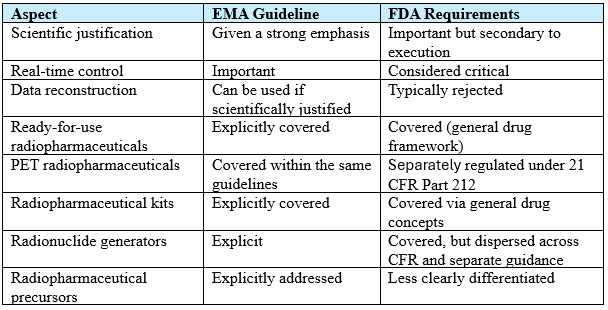
Comparison With FDA Approaches
The EMA guideline provides a unified approach to radiopharmaceuticals. This contrasts with the more fragmented framework used by the FDA, which comprises:
- 21 CFR Part 212: cGMP for PET drugs only
- 21 CFR Part 315: Diagnostic radiopharmaceuticals
- Plus, additional guidance documents for:
- microdose diagnostic radiopharmaceuticals
- oncology therapeutic radiopharmaceuticals
- PET drug oversight, stability, sterility pending release, etc.
Overall, the FDA documentation is more rule‑based and operationally prescriptive, whereas the EMA approach is technical, providing more scope for manufacturers to adopt different pathways. In summary, the main differences are:
Summary
The draft EMA guideline represents a major modernization of EU regulatory expectations for radiopharmaceutical quality. In particular, the guideline recognizes the practical realities of short half‑lives, on‑site preparation, and rapid release, while reinforcing the need for robust upstream control, process validation, and scientifically justified specifications.
For manufacturers, it provides clearer alignment with European Union GMP, the European Pharmacopeia, and ICH principles (as interpreted through a radiopharmaceutical‑specific lens). For regulators and inspectors, the text establishes a consistent and transparent framework for assessing quality across what is becoming an increasingly diverse radiopharmaceutical landscape.
References:
- European Medicines Agency. Guideline on quality of radiopharmaceuticals (draft). EMA/CHMP/QWP/363827/2025, Committee for Medicinal Products for Human Use (CHMP), 2025: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-radiopharmaceuticals-revision-2_en.pdf
- Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. Current consolidation version dates 01/01/2025. http://data.europa.eu/eli/dir/2001/83/oj
- ICH Topic M 4 Q. Common Technical Document for the Registration of Pharmaceuticals for Human Use – Quality: Quality Overall Summary Of Module 2 And Module 3. CPMP/ICH/2887/99, July 2003: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-m4q-common-technical-document-registration-pharmaceuticals-human-use-quality-step-5_en.pdf
- Allisy, A. From the curie to the becquerel, Metrologia, 1995; 32 (6): 467–479 doi:10.1088/0026-1394/31/6/006
- Brans B, Bodei L, Giammarile F, et al. Clinical radionuclide therapy dosimetry: the quest for the "Holy Gray". Eur J Nucl Med Mol Imaging. 2007;34(5):772-786. doi: 10.1007/s00259-006-0338-5
- Neels O, Patt M, Decristoforo C. Radionuclides: medicinal products or rather starting materials? EJNMMI Radiopharm Chem. 2019;4(1):22. doi: 10.1186/s41181-019-0074-3
- de Roode KE, Joosten L, Behe M. Towards the Magic Radioactive Bullet: Improving Targeted Radionuclide Therapy by Reducing the Renal Retention of Radioligands. Pharmaceuticals (Basel). 2024;17(2):256. doi: 10.3390/ph17020256
- Edem PE, Fonslet J, Kjær A et al, In Vivo Radionuclide Generators for Diagnostics and Therapy. Bioinorg Chem Appl. 2016;2016:6148357. doi: 10.1155/2016/6148357
About The Author:
 Tim Sandle, Ph.D., is a pharmaceutical professional with wide experience in microbiology and quality assurance. He is the author of more than 30 books relating to pharmaceuticals, healthcare, and life sciences, as well as over 170 peer-reviewed papers and some 500 technical articles. Sandle has presented at over 200 events and he currently works at Bio Products Laboratory Ltd. (BPL), and he is a visiting professor at the University of Manchester and University College London, as well as a consultant to the pharmaceutical industry. Visit his microbiology website at https://www.pharmamicroresources.com.
Tim Sandle, Ph.D., is a pharmaceutical professional with wide experience in microbiology and quality assurance. He is the author of more than 30 books relating to pharmaceuticals, healthcare, and life sciences, as well as over 170 peer-reviewed papers and some 500 technical articles. Sandle has presented at over 200 events and he currently works at Bio Products Laboratory Ltd. (BPL), and he is a visiting professor at the University of Manchester and University College London, as well as a consultant to the pharmaceutical industry. Visit his microbiology website at https://www.pharmamicroresources.com.