
Can We Eradicate Tech Transfer And Other 20th Century Pharma Manufacturing Practices?
By Mark F. Witcher, Ph.D., biopharma operations subject matter expert
While warp-speed manufacturing is a pandemic term, the concept is an important part of the FDA’s 21st century vision of “a maximally efficient, agile, flexible pharmaceutical manufacturing sector that reliably produces high-quality drugs without extensive oversight.”1 Reaching 21st century performance will require replacing many of the industry’s 20th century practices by building new and innovative methods and capabilities to rapidly, efficiently, and reliably develop and license new products, including therapeutics and vaccines to mitigate and control pandemics. This article briefly describes how new innovative nonprescriptive paradigms can be used to replace current outdated approaches to better reach the FDA’s 21st century vision.
“a maximally efficient, agile, flexible pharmaceutical manufacturing sector that reliably produces high-quality drugs without extensive oversight.”1 Reaching 21st century performance will require replacing many of the industry’s 20th century practices by building new and innovative methods and capabilities to rapidly, efficiently, and reliably develop and license new products, including therapeutics and vaccines to mitigate and control pandemics. This article briefly describes how new innovative nonprescriptive paradigms can be used to replace current outdated approaches to better reach the FDA’s 21st century vision.
In the 20th century, the pharmaceutical industry created and used approaches to deal with very primitive biopharmaceutical manufacturing technologies (i.e., low yields, specially designed stainless-steel equipment in single product facilities, etc.). These costly and inefficient approaches greatly limited the industry’s ability to commercialize new products.
To protect patients, regulatory agencies developed policies and methods that emphasized compliance with good manufacturing practices (GMPs) for approving new products and regulating an unsophisticated industry experiencing a wide range of product quality problems as it struggled with new, very complex products and processes. However, as medical, biopharmaceutical, and manufacturing technologies such as single-use equipment and much higher yields continued to advance, much of the industry’s engineering and management did not evolve and implement new methods for dealing with the challenges or take advantage of the many opportunities created by the new technologies.
For reasons that are not clear and beyond the scope of this article, the pharmaceutical industry has become obsessed with compliance with regulatory guidelines to the exclusion of continuing to evolve better engineering and management approaches. To make matters worse, the industry for many years blamed the slow and extremely expensive development of new products on regulatory constraints rather than seeking to improve their own methods. As a consequence, the FDA developed programs such as expedited review, etc. to minimize its impact on the critical path for new products. While the FDA’s focus on science has allowed it to adapt in many ways to the advancing technologies, its limited engineering expertise has resulted in a compliance-centric and performance-based regulatory strategy rather than fostering and encouraging advanced engineering approaches. Regardless of the history, the industry and its regulatory agencies must aggressively develop and support the evolution of innovative engineering tools and operational paradigms for developing new products if the many opportunities of 21st century technologies are to be realized.
While most industries have competitive forces making speed and efficiency essential for survival, the pharmaceutical industry is challenged to develop highly efficient and agile methods without the motivation of a constant threat of being surpassed by a competitor. Regulatory agencies and some in the industry are trying to install artificial competitive forces that might help. But to a large extent, the industry must self-initiate efforts to address society’s health challenges without the assistance of competitive forces.
Because of the need to clearly demonstrate the safety and efficacy of its products, the pharmaceutical industry must use a phased approach for developing products. Starting with medical research, a company identifies a product, develops a process to make the product, and then scales up the manufacturing process as additional material is required for preclinical and clinical testing and, if all goes well, commercial manufacturing. This approach has led to many organizational siloes that have greatly hindered the product development effort by adding time, cost, and many risks.
A good example of 20th century thinking is readily accepting the significant time losses and increased costs of transferring production technologies between different groups, either internally or externally, to gain access to critical manufacturing resources to support the next development phase.
Eliminating The Need For Tech Transfer
Manufacturing technology is transferred only when the current capabilities cannot handle the next development phase, thus requiring a change in resources, almost always to remove a deficit in manufacturing capability. As a product moves through its development life cycle, it becomes increasingly difficult to transfer the technology from one organization to another because of the increasing complexity and sophistication of the manufacturing and quality control systems.
The challenges and significant increases in time delays of transferring pharmaceutical technology are well summarized in a report published by McKinsey & Co.2 Tech transfer is a complex nonvalue-added activity that adds costs, time, and significant risks, especially to the product’s successful development.3 The critical path for developing any new products should go primarily through the preclinical and clinical trials required for establishing safety and efficacy. Given advances in clinical trial designs and regulatory approval mechanisms, particularly emergency use authorization (EUA), keeping development and manufacturing off the critical path has become very difficult.
The challenges of providing very expensive manufacturing resources for new products is daunting. The product may fail at any phase of development, either making a specialized facility obsolete or requiring the manufacturing facility to quickly remove the failure to make room for other products or new candidates. Sizing a facility is difficult because capacity requirements for preclinical, clinical, and commercial manufacturing can vary by several orders of magnitude because of uncertainty in process yields, dose requirements, and patient populations. Additional uncertainty can also come from the number and nature of the unit operations required to make the product. For example, if continuous manufacturing options are being considered, it might be necessary to simultaneously develop more than one process or process segment for an important product. To prevent the necessity of transferring the technology, the manufacturing resources must have the agility and adaptability to meet the above challenges while under a great deal of pressure to produce material as rapidly as possible.
Meeting this challenge requires making smart investments in manufacturing resources capable of supporting all the manufacturing requirements of the product from development through launch. Understanding 21st century manufacturing begins by understanding the three elements of a manufacturing enterprise.
A 21st Century Manufacturing Enterprise
Pharmaceutical manufacturing requires three elements: 1. The manufacturing process that produces the product; 2. The facility that contains and protects the manufacturing process; and 3. The control infrastructure, frequently call the quality management system (QMS), that controls the operation of the manufacturing processes within the surrounding facility.4,5 The three elements shown in Figure 1 must work as a single, well-coordinated unit if the manufacturing process is to be protected from a wide variety of risks and supported by the facility while operationally controlled by the infrastructure.6
![]()
Figure 1: The manufacturing enterprise – The manufacturing process is operated within the facility under the control of the QMS.
The key to achieving the necessary agility and adaptability is to simultaneously operate a wide variety of different manufacturing processes within the facility under the control of a single quality management infrastructure. The essential feature is a facility layout that can completely segregate one product from other products while allowing all processes to be simultaneously operated without interfering with each other. In addition, if the unit operations for a product can be subdivided into separate operating entities, then they can be placed within the facility in a variety of configurations and locations to achieve additional operating flexibility. The facility layout must also provide the necessary process connectivity to efficiently operate the entire process for every product. Finally, if support functions, such as media and buffer preparation and storage, are treated as independent unit operations, then they can also be added, deleted, or scaled as necessary. Such a facility can quickly adapt to a wide variety of different processes for making many different products at different scales and capacities.
A facility designed with both bi- and unidirectional resource flows within a highly segregated cleanroom matrix, such as the multipurpose facility (MPF), provides the ability to independently operate many different processes, allowing products to move in and out of the enterprise depending on each product’s life cycle manufacturing needs.7,8
An MPF based manufacturing enterprise has the following capabilities:
-
Multiproduct/multiphase – It can simultaneously manufacture a wide variety of biologics, vaccines, and therapies using a wide range of process formats at different scales and capacities to support a product from process development through commercial launch.7,8
-
Adaptable – It can efficiently increase or shrink facility resources to utilize only the resources required for a product, thus freeing valuable assets for other products or manufacturing requirements.
-
Agility (e.g., fast flexibility) – It can add or remove process unit operations in hours or days without impacting ongoing manufacturing operations.
-
Support process development and validation – It provides an environment for collecting process information to build and expand a well-structured design space as the process is scaled up from development through commercial launch.
-
Support process evolution – It provides for continuous evolution of a process’ procedures, practices, and control strategies as the product transitions from development through Phase 3 and manufacturing launch.
-
Priority driven – It is able to quickly remove failures, add new candidates, expand successes, and remove low-priority products to manufacture high-priority products.
-
Personnel continuity – It supports using the same operating personnel and their knowledge and experience for all phases of a product’s development.
-
Minimize regulatory oversight requirements – By using the same QMS operating systems and facility resources for all products, the MPF can be a “known manufacturing asset” to regulatory agencies, perhaps even described by a “facility master file.” Once reviewed and inspected, products could be added or removed with minimal regulatory oversight.5
A manufacturing enterprise with such capabilities can be very efficient by having a very high utilization rate and eliminating the need for tech transfer. However, operating an MPF requires understanding and integrating a large number of processes and must effectively control a wide variety of threats and risks that can impact the enterprise’s many processes and systems.6
Structuring Processes To Manage Risks
Designing and operating the enterprise’s interacting network of processes and support systems is very challenging. The primary requirement of pharmaceutical manufacturing is controlling risks to assure reliability and consistent product quality.9 The industry does not currently have a method for systematically identifying, analyzing, controlling, and, most importantly, communicating the wide variety of risks the manufacturing enterprise must control.10 However, viewing the entire manufacturing enterprise, including the manufacturing process itself, as a sequence or network of individual processes enables the use of a simple risk-based approach for designing, testing, and operating all the enterprise’s systems, including the manufacturing process sequence of unit operations.
As shown in Figure 2, a risk analysis can be carried out for a process by viewing a risk as an output risk consequence that might result from a possible input threat successfully passing through the process.11 This basic risk element can be used to analyze and manage the risks associated with both a very simple process as well as complex systems. Bioreactors, procedures, practices, material flows, etc. are all systems composed of processes and subprocesses. Using this approach, all the enterprise’s systems can be collectively designed to prevent a wide variety of threats from resulting in negative consequences to the product.
![]()
Figure 2: A basic risk element for managing risks – All risks can be viewed as a possible threat of likelihood LT entering a process with a likelihood LP of producing a risk consequence of severity CS and likelihood of LC. If all inputs are viewed as possible threats that might produce a negative risk consequence, a risk management exercise is possible by assessing the severity and likelihoods of both the threats and consequences.12
If the basic element in Figure 2 is used to analyze the various systems of processes using system risk structures, then many risk management exercises can be carried out using the ICH Q9 risk method summarized in Figure 3.
![]()
Figure 3: System Risk Structure Analysis – The four stages defined in ICH Q9 provide an effective method of evaluating the risks defined by a system risk structure (SRS) and documented using the SRS and a risk register (RR).12
As shown, the risks are initially identified as input threats and output consequences of concern. The threats and consequences are then structured to establish their causal relationships by connecting the inputs and outputs via processes to make a process flow diagram (PFD). The PFD is then turned into a system risk structure (SRS) by assessing the severity and likelihood of the threats and consequences per the basic risk element shown in Figure 2. Unacceptable risks are then further evaluated to gain additional information and understanding for acceptance or mitigation. Unacceptable risks are then controlled by adding or modifying processes in the SRS to reduce the likelihood of important consequences occurring so they can be accepted.
The SRS and the risk register (RR) are used to reach alignment and consensus on the design of the processes that make up the system. Effective risk communication between the pharmaceutical companies and regulators is perhaps the most essential element in minimizing the regulatory oversight component of the FDA’s vision. The SRS and RR provide the rationale for effective communication and alignment of the many risks with regulatory requirements.
However, assessing risks must happen within an overall approach for developing and operating the processes. Every process in all three manufacturing enterprise elements has a life cycle that can be described by four questions: What? How? Will it work? and Did it work?13 The answers to the four questions define the process’ life cycle.
For pharmaceutical manufacturing, all four questions must be answered, and the answers must be documented to support regulatory approval. As shown in Figure 4, the life cycle of every process from bioreactor to procedures can be managed using a lifecycle process development & validation (LPDV) paradigm.14 In addition, the risks associated with each process, including the connections between all the processes can be managed using an SRS approach as shown in Figures 2 and 3.12
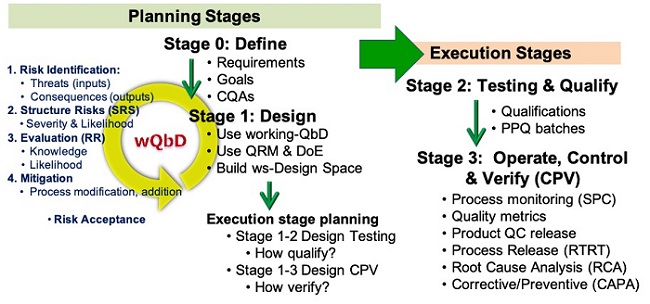
Figure 4: Life cycle Process Development & Validation – The two planning stages define and design the process prior to the two execution stages.
Because of the ever-compounding complexity of the products and the processes required to make them, using a universal process development and management approach for all processes that facilitates the use of a straightforward risk management tool can greatly simplify the way manufacturing enterprises, including the manufacturing process unit operations, are developed, validated, and operated.
Conclusion
While the references cited in this article represent a set of ideas and concepts, they are far from the last word on the capabilities needed to achieve the FDA’s 21st century vision. The industry can and must continue to evolve these as well as conceive new ideas to make product development more efficient for quickly developing novel high-quality products to meet the many medical needs.
In the long run, the 21st century approaches must be based on concepts that are independent of the technologies, along with intrinsically producing clear rationales derived from fundamental risk analysis and mitigation tools used as part of a straightforward nonprescriptive process life cycle development and validation paradigm.
Regulatory agencies can facilitate and support this evolution by using the same approach in the next generation of regulatory guidelines for ICH Q8, Q9, Q10, and the all-important next generation of the FDA’s 2011 Principles of Process Validation to include complete life cycle management of all the processes required for manufacturing pharmaceuticals.
References:
- FDA – Pharmaceutical cGMPs for the 21st century – a risk-based approach. Sept 2004.
- McKinsey & Company, “Why tech transfer may be critical to beating COVID-19, July 23, 2020. https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/why-tech-transfer-may-be-critical-to-beating-covid-19
- Witcher, M. F., “Phase III Clinical Trials – Ever Wonder Why Some Products Unexpectedly Fail? ISPE iSpeak Blog Pharmaceutical Engineering, Aug. 7, 2019. https://ispe.org/pharmaceutical-engineering/ispeak/phase-iii-clinical-trials-ever-wonder-why-some-products-unexpectedly-fail
- Witcher, M. F., “Impact of Facility Layout on Developing and Validating Segregation Strategies in the Next Generation of Multi-product, Multi-phase Biopharmaceutical Manufacturing Facilities;” Supplement to Pharm. Engr.; January 2014. https://ispe.org/pharmaceutical-engineering/ispeak/how-does-facility-layout-affect-segregation-strategies-nextgen
- Witcher, M. F. “How FDA’s 21st Century Goals can be realized by using a Multi-purpose Manufacturing Facility,” Pharmaceutical Technology, 2018. http://www.pharmtech.com/how-fda-s-21st-century-goals-can-be-realized-using-multi-purpose-manufacturing-facility-0?pageID=2
- Witcher, M. F. and H. Silver “Comparing Facility Layout Options for Managing Business and Operating Risks” BioPharm International 32 (6) 2019. http://www.biopharminternational.com/comparing-facility-layout-options-managing-business-and-operating-risks?pageID=1
- Witcher, M. and H. Silver, "Multi-Purpose Biopharmaceutical Manufacturing Facilities Part 1: Product Pipeline Manufacturing," Pharmaceutical Technology 42 (9) 2018. http://www.pharmtech.com/multi-purpose-biopharmaceutical-manufacturing-facilities-part-1-product-pipeline-manufacturing?pageID=1
- Witcher, M. and H. Silver, "Multi-Purpose Biopharmaceutical Manufacturing Facilities Part 2: Large-Scale Manufacturing," Pharmaceutical Technology 42 (11) 2018. http://www.pharmtech.com/multi-purpose-biopharmaceutical-manufacturing-facilities-part-ii-large-scale-production
- FDA - ICH Q9 – Quality Risk Management
- Witcher, M. F., “Stop Talking about Risk, Get Serious about Developing Effective Risk Management Tools,” ISPE Pharmaceutical Engineering iSpeak Blog, 19 June 2019 https://ispe.org/pharmaceutical-engineering/ispeak/get-serious-about-developing-effective-risk-management-tools?
- Witcher MF. Analyzing and managing biopharmaceutical risks by building a system risk structure (SRS) for modeling the flow of threats through a network of manufacturing processes. BioProcess J, 2017; 16. https://doi.org/10.12665/J16OA.Witcher
- Witcher, M. F., System Risk Structures: A New Framework for Avoiding Disasters by Managing Risk, BioProcess Online, July 13, 2020. https://www.pharmaceuticalonline.com/doc/system-risk-structures-a-new-framework-for-avoiding-disaster-by-managing-risks-0001
- Witcher MF. Integrating development tools into the process validation lifecycle to achieve six sigma pharmaceutical quality. BioProcess J, 2018; 17. https://doi.org/10.12665/J17OA.Witcher.0416
- Witcher, M. F., A Functional History of Process Validation: Part 2 – The Key To A More Effective Future, BioProcess Online, August 2020. https://www.pharmaceuticalonline.com/doc/a-functional-history-of-process-validation-part-the-key-to-a-more-effective-future-0001
About The Author:
 Mark F. Witcher, Ph.D., has over 35 years of experience in biopharmaceuticals. He currently consults with a few select companies. Previously, he worked for several engineering companies on feasibility and conceptual design studies for advanced biopharmaceutical manufacturing facilities. Witcher was an independent consultant in the biopharmaceutical industry for 15 years on operational issues related to: product and process development, strategic business development, clinical and commercial manufacturing, tech transfer, and facility design. He also taught courses on process validation for ISPE. He was previously the SVP of manufacturing operations for Covance Biotechnology Services, where he was responsible for the design, construction, start-up, and operation of their $50-million contract manufacturing facility. Prior to joining Covance, Witcher was VP of manufacturing at Amgen. You can reach him at witchermf@aol.com or on LinkedIn.
Mark F. Witcher, Ph.D., has over 35 years of experience in biopharmaceuticals. He currently consults with a few select companies. Previously, he worked for several engineering companies on feasibility and conceptual design studies for advanced biopharmaceutical manufacturing facilities. Witcher was an independent consultant in the biopharmaceutical industry for 15 years on operational issues related to: product and process development, strategic business development, clinical and commercial manufacturing, tech transfer, and facility design. He also taught courses on process validation for ISPE. He was previously the SVP of manufacturing operations for Covance Biotechnology Services, where he was responsible for the design, construction, start-up, and operation of their $50-million contract manufacturing facility. Prior to joining Covance, Witcher was VP of manufacturing at Amgen. You can reach him at witchermf@aol.com or on LinkedIn.