
Certifying Pharmaceutical Exports: A Roadmap To EMA's Certificate Of Medicinal Product
By Michael Cooper, Clinical and Regulatory Affairs, Pharmatech Associates
In Part 1 of this two-part series, we discussed requirements for exporting U.S.-manufactured material to foreign markets supported by a certificate of pharmaceutical product (CPP) issued by the FDA. Here in Part 2, the reader will learn about exporting EU-manufactured product to foreign markets leveraging a certificate of medicinal product (CMP) issued by the European Medicines Agency (EMA). We will discuss obtaining an EMA-issued CMP for a product not approved in the EU, under a provision known as Article 58.
supported by a certificate of pharmaceutical product (CPP) issued by the FDA. Here in Part 2, the reader will learn about exporting EU-manufactured product to foreign markets leveraging a certificate of medicinal product (CMP) issued by the European Medicines Agency (EMA). We will discuss obtaining an EMA-issued CMP for a product not approved in the EU, under a provision known as Article 58.
Before proceeding into how the CPP is administered in the European Economic Area (EEA) — where it is known as a certificate of medicinal product, or CMP — it is necessary to understand how marketing authorizations for medicinal products work within the EEA.
For memory, while the European Union (EU) is comprised of 27 member states, three additional countries comprise the EEA and follow the same pharmaceutical regulations: Norway, Iceland, and Liechtenstein. The European Medicines Agency (EMA) is the health authority, and the reviewing body is the Committee for Medicinal Products for Human Use (CHMP).
Within the EEA, medicinal products may be authorized through one of four pathways as described in Table 1:
Table 1: Approval Pathways for a Marketing Application Authorization (MAA)
|
Description |
Pros |
Cons |
|
National Authorization Procedure |
Can select member state supportive of new technology (e.g., gene therapy) Most common for generics and OTCs Mutual recognition procedure can follow |
Authorization limited to single member state |
|
Mutual Recognition Procedure |
Abbreviated review in other member states |
Potential for differences in opinion, requiring resolution by CHMP |
|
Decentralized Approval |
Simultaneous submission to multiple member states when criteria for Centralized Approval not met |
Potential for differences in opinion, requiring resolution by CHMP |
|
Centralized Approval |
Simultaneous access to all EEA markets |
Reimbursement negotiated with each member state |
Human and veterinary medicinal products that meet any of the following criterion require centralized approval:
- All medicinal products derived from biotechnology; advanced therapy medicinal products (a medicine derived from gene therapy, cell therapy, or tissue engineering, also referred to as ATMP)
- Medicinal products for human use containing new active substance for important indications (e.g., cancer, diabetes, and viral diseases)
- Orphan medicinal products
- Medicinal products for veterinary use intended primarily for use as performance enhancers, to promote the growth of treated animals, or to increase yields from treated animals
During review of a centralized MAA, the EMA will assign a lead technical reviewer (rapporteur) and a supporting technical reviewer (co-rapporteur). The rapporteurs are selected based on their expertise in the therapeutic area from a member state’s national medicine regulatory authority (NMRA). A rapporteur is also assigned from the Pharmacovigilance Risk Assessment Committee (PRAC). The scope of this article is limited to (1) the process of applying for a CMP for a centrally authorized medicinal product, and (2) the process of applying for a CMP for a medicinal product that is not authorized in any EEA country under Article 58. Each member state’s NMRA will have its own regulations on how to request a CMP for a nationally authorized medicinal product, so this scenario will not be covered.
Similar to a CPP issued by the FDA, an EMA-issued CMP certifies that the quality, safety, and efficacy (QSE) of the assessed product meets stringent requirements and that associated facilities meet current good manufacturing practice (cGMP) requirements. As a result, patients in new markets can gain quicker access to important medicines. Note that the EMA can only issue a CMP if all associated manufacturing and batch release sites are in good GMP standing based on their most recent inspections.
CMPs For Centrally Authorized Products
The CMP incorporates the WHO format and recommendations, as discussed in part one of this two-part series. It can be issued in any of the 24 official languages of the EU. The certificate template will include information in English, Spanish, French, and Portuguese. Product information will be presented in the requested language, whereas the accompanying European Public Assessment Report—EPAR, also known as a Summary Basis of Approval in other markets—will be issued in English.
Generally, CMPs are requested by the marketing authorization holder (MAH) through a business address within the EEA. A partner can request the CMP on their behalf with a permission letter. The letter must originate from the MAH and be provided with the request for certificates. It must be sent for every product requested on behalf of the MAH. It will be kept on file and the requester will be asked to declare in the application form that the last submitted permission letter is valid.
Many rest-of-world countries have strong requirements and preferences for legalization of CMPs (e.g., apostille, etc.). Because the EMA agrees with the WHO that certifications are not necessary, NMRAs making this request should contact the EMA directly through the Procedures Office, Committees and Quality Assurance Department by mail, courier, or email at certificate@ema.europa.eu. However, in my experience, many sponsors will take on legalization themselves to expedite submission in importing countries.
The EMA can provide CMPs as an electronic certificate or as a printed certificate:
- The authenticity of electronic certificates is assured by an electronic signature that complies with the electronic Identification, Authentication, and Trust Services (eIDAS) regulation, Regulation (EU) No. 910/2014. Note that not all countries (e.g., Latin America region) accept electronic certificates at this time.
- Paper certificates are printed on A4-size yellow certificate paper with a colored EMA logo, stamped with an original EMA stamp, and signed by an authorized person. All pages attached to the certificates have a printed EMA stamp and are glued to the certificates with a blue binder strip.
There is no expiry date assigned to a CMP, as it reflects the EU marketing authorization on the date of CMP issue.
The EMA issues CMPs as a set, which is six identical original CMPs with a unique marketing authorization number, addressed to the same importing country, issued in the same official language of the European Union, and having identical annexes. Refer to Figure 1 for a description of fees and time frames.

Figure 1: Fees and timelines for CMP requests
CMPs For Medicinal Products Not Authorized In The EEA — Article 58
For products not approved in the EEA, CMPs can be requested under Article 58 of Regulation (EC) No. 726/2004 (Article 58). Applicants should notify the EMA of their intention seven months prior to the intended submission date so that rapporteurs (reviewers) can be assigned. There are two steps to this process: (1) requesting eligibility for a CMP under Article 58 (there is no charge), and (2) requesting the actual CMP.
The EMA recommends requesting scientific advice when applying for eligibility under Article 58. The cost is €44,400 to €89,000, although it is free for pediatric questions. Fee reductions are available for small and midsize enterprises (SMEs). The timeline is 40 days, unless a meeting is required, in which case it is 70 days.
Table 2 below describes products eligible for CMPs under Article 58.
Table 2: Products Eligible for Article 58 CMPs
|
Description |
Comments |
|
Vaccines |
|
|
WHO Expanded Program on Immunization |
Diphtheria, whooping cough, tetanus, measles, poliomyelitis, tuberculosis, hepatitis B, and Hemophilus influenzae type b (Hib); Pneumococcal conjugate vaccine and rotavirus vaccines are being added. |
|
WHO public health priority disease series 1:
|
Many treatments already ineffective or will soon be. |
|
WHO series 2:
|
Treatments exist but improvements needed (e.g., pediatric dosages of cancer medicines, side effects of antidepressants, oxytocin and insulin are not heat-stable, etc.) |
|
WHO series 3:
|
Treatment does not yet exist or inadequate; basic research needed to establish biomarkers. |
|
WHO series 4:
|
Treatment does not yet exist or inadequate |
|
WHO-managed stockpile for emergency response |
|
|
Medicinal Products |
|
|
WHO target diseases |
HIV/AIDS, malaria, tuberculosis, lymphatic filariasis (elephantiasis), trachoma, leishmaniasis, schistosomiasis, African trypanosomiasis (sleeping sickness), onchocerciasis (river blindness), dengue fever, Chagas disease, leprosy, and intestinal helminths |
In addition to the products defined above, eligible products may include new formulations, new pharmaceutical forms, or new routes of administration for products already authorized in the EU, as well as fixed-dose combination products and generic products.
The process for requesting a CMP under Article 58 is described in Figure 2 below:
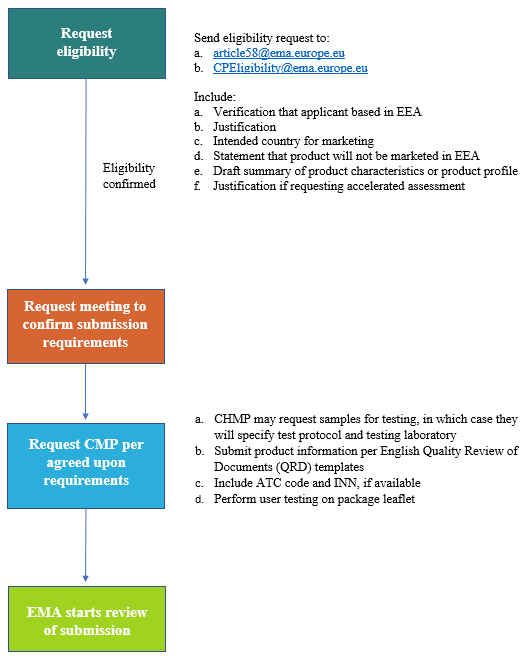
Figure 2: Process for requesting a CMP under Article 58
The review timeline for an Article 58 CMP is the same as for an MAA. Review starts approximately two weeks after the defined monthly submission date on the EMA website.
Once the clock starts, you are at day 1. The clock stops at day 120 and day 180, when a list of questions (LoQ) and outstanding issues (LoOI) are received from the agency. The clock restarts once the applicant submits responses. At day 180, the agency may request that the applicant answer questions in a face-to-face meeting. A CHMP scientific opinion (approval or rejection) is issued by day 210.
Applicants may request an accelerated evaluation under Article 58, which will be jointly evaluated by CHMP and the WHO.
Refer to Figure 3 below for significant milestones in the review process.

Figure 3: Evaluation timeline for CMP request under Article 58
After CHMP adopts the scientific opinion, it and associated annexes are sent to relevant stakeholders. Within two months, an EPAR is prepared with WHO and published on the EMA website.
Table 3 provides a comparison of MAA and Article 58 CMP requirements:
Table 3: Comparison of MAA and Article 58 CMP Application Processes
|
Requirement for a Centralized MAA |
Requirement applies to Article 58 CMP application? |
|---|---|
|
Applicant notifies the EMA of intent to submit application and proposed month at least 7 months prior |
Yes |
|
(Co-)Rapporteurs assigned from member states to lead technical assessment, based upon their expertise in therapeutic area |
Yes |
|
The package leaflet undergoes user testing per Directive 2001/83/EC to ensure that all information is easily understood |
Yes |
|
Application follows the EMA’s defined start-of-review dates based upon submission date |
Yes |
|
An environmental risk assessment (ERA) is in place |
Recommended but not required |
|
The CHMP issues a scientific opinion on the application within 210 days, with start/stop of the review clock for the applicant to respond to agency questions (in writing or orally) and to resolve outstanding issues. |
Yes |
|
A pharmacovigilance system and risk management plans (RMPs) are in place for the product |
Yes |
With a 210 working day review period for an Article 58 CMP request (not counting time spent responding to agency questions), this is not a particularly fast process for supporting overseas registration.
Conclusion
We have reviewed how a product approval within the EEA can be leveraged to support registration in new markets through the CMP. For products not approved in the EEA, sponsors can still request a CMP under Article 58. While the described process may sound laborious, sponsors interested in this pathway should request an accelerated assessment in order to meet their business timelines.
Resources:
- Information package for certificates of medicinal products issued by the European Medicines Agency (EMA), 30 March 2020, EMA/119843/2013 Rev 15, Parallel Distribution and Certificates: https://www.ema.europa.eu/en/documents/other/information-package-certificates-medicinal-products-issued-european-medicines-agency_en.pdf
- Certification of medicinal products: https://www.ema.europa.eu/en/human-regulatory/post-authorisation/certification-medicinal-products
- EMA procedural advice for medicinal products intended exclusively for markets outside the European Union under Article 58 of Regulation (EC) No 726/2004 in the context of co-operation with the World Health Organisation (WHO). August 2017, EMA/534107/2008 Rev.1: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/european-medicines-agency-procedural-advice-medicinal-products-intended-exclusively-markets-outside/2004-context-cooperation-world-health_en.pdf
About The Author:
 Michael Cooper is a clinical and regulatory affairs program manager at Pharmatech Associates. He has over 20 years of experience in the biopharmaceutical industry, with expertise in regulatory affairs chemistry, manufacturing, and controls (CMC) submissions; GMP inspections for biologics and vaccines; QA lot release of drug substance and drug product; deviation and CAPA resolution; and facilities, utilities, and equipment validation.
Michael Cooper is a clinical and regulatory affairs program manager at Pharmatech Associates. He has over 20 years of experience in the biopharmaceutical industry, with expertise in regulatory affairs chemistry, manufacturing, and controls (CMC) submissions; GMP inspections for biologics and vaccines; QA lot release of drug substance and drug product; deviation and CAPA resolution; and facilities, utilities, and equipment validation.