
Draft FDA Labeling Guidance Proposes More Clarity For Providers
By Alexander K. Aust, M.S., MBA, Aust Business Solutions

The FDA's draft guidance on labeling for prescription drugs and biologics focuses on giving healthcare practitioners clear and precise information that they can understand.
The draft, titled Dosage and Administration Section of Labeling for Human Prescription Drug and Biological Products — Content and Format, is to replace the original guidance of the same name published in 2010. The agency released the draft in January and withdrew the 2010 guidance of the same name. A public comment window for the revised draft closes March 14.
The FDA published the original guidance to direct drugmakers on the best ways to format information on labels for pharmaceutical and biopharmaceutical products. Since then, the biologics space has exploded with activity, so it makes sense that the agency would revisit its labeling standards and provide more clarity on its expectations for prioritizing information and organizing it with headings, subsections, tables, and cross-references to other sections. In the draft guidance, FDA provides more examples of dos and don’ts that have become more relevant since the first version.
In general, the FDA recommends not cluttering up the section with information considered to be general medical knowledge or broad information that does not specifically pertain to the drug being considered. This is because such statements serve as a distraction from the critical information that is actually recommended on the label.
Agency Draws A Line Between ‘Dose’ and ‘Dosage’
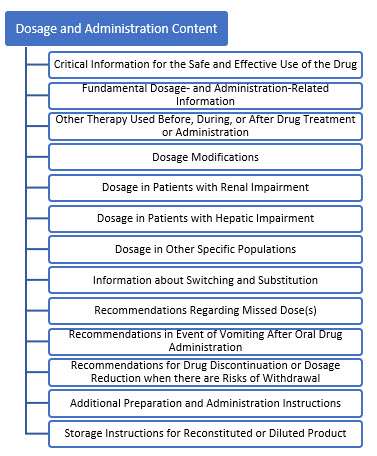
The draft guidance notes a key difference between the definitions of “dose” and “dosage.” It defines dose as “a specific amount of drug taken at one time” and it defines dosage as “a specific amount of drug administered at a specific frequency (and over a certain duration, if applicable). The appropriate term (dosage versus dose) should be used throughout the labeling depending on the information being discussed.” This differentiation is applicable to the Dosage and Administration Section of Labeling for Human Prescription Drugs and Biological Drugs. The draft guidance breaks down FDA’s content recommendations as displayed in Figure 1.
Figure 1. Content Sections of January 2023 Draft Guidance, Dosage and Administration Section of Labeling for Human Prescription Drug and Biological Products
Draft Guidance Acknowledges Greater Complexity Of Biologics
The 2023 Draft Guidance provides more specific examples for biologics, as they are typically more complex in formulation, dosage, and administration than traditional human prescription drugs. The increased complexity in the preparation and administration of biologics is because most of them are parenteral drug products (sterile injectables). Biologics are increasingly more complicated and require additional information to be included for the safe and effective use of the drug. As such, particular attention needs to be placed on when and where this critical information is presented.
Typically, this information comes before the “fundamental dosage- and administration-related information.” Often, biologics do not get to enjoy the simplicity of room temperature storage as a long-term storage solution. The drug is simply not stable enough to be stored at 20 to 25 degrees C and will require refrigeration or freezing. The cold chain custody requirement of biologics is why FDA provides specific instructions and examples for how to prepare a product for administration that is stored in the refrigerator or freezer. The majority of biologics are filled into vials, and the product is withdrawn with a syringe. The draft guidance provides information about when to be specific when it comes to considering the materials of construction for the containers and devices needed for administration, while advising to be more generic when it comes to listing specific manufacturers.
Essentially, FDA advises against displaying brand loyalty when it comes to vials and syringes. When discussing diluents to be used for reconstitution, be as specific as you can be. This means that FDA wants to see the name and strength of the diluent used. The examples in the draft guidance are: “use ‘0.9% Sodium Chloride Injection’ instead of ‘normal saline’ or ‘saline’”. Additionally, if the drug requires filtration prior to administration, then FDA recommends, “this section should identify the appropriate filter(s) and pore size (e.g., low-protein binding, 0.2 micron, in-line filter).”
A distinction not included in the 2010 guidance is the recommendation to provide additional consideration for intramuscular or subcutaneous injections. For instance, “If the injection depth is important for administration or the injection duration is lengthy (e.g., two minutes or longer), include the recommended injection depth or the recommended duration of the injection, respectively.” Also, FDA provides additional information in the draft guidance for system dosage forms (i.e., a delivery system that governs the rate of release of the drug); “this section should provide the rate of release and the total duration of the drug release, and instructions for application, rotation and removal when applicable.”
Old And New Drafts Both Emphasize The Importance Of Visual Inspections
One thing that hasn’t changed between the 2010 and 2023 versions is that a parenteral must include the following statement in the Dosage and Administration section: “Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.” Visual inspection can be difficult to perform with an amber vial if the drug is sensitive to light. It is also likely that liposome products will be difficult to inspect because of their milky (non-clear) coloration due to the functional excipients encapsulating the drug substance.
Focus On Liposome Drug Products Follows An Innovation Surge
It is telling how much progress has been made in drug delivery systems (DDS) since 2010, because FDA includes an additional example for the effective and safe use of the drug. This is also evident by the 2023 draft referencing the April 2018 guidance for industry, Liposome Drug Products – Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation, for insight into how to handle drug products that utilize lipid-based nanocarriers. Here’s what the 2018 guidance has to say about label instructions for liposome drug products:
“Where appropriate, reconstitution instructions and a statement regarding the appropriate in-use period should be provided. This information should be provided for both unloaded liposomes that are reconstituted with a drug substance-containing solution at the time of use and for products in which the drug substance is loaded into the liposomes during manufacturing. For liposome drug products that are labeled for use after mixing with other approved drug products (e.g., large volume injectable solutions), admixing instructions and a statement regarding the appropriate in-use period of the admixed product should be included/ As warranted, include storage conditions for the reconstituted drug, robustness of the liposome drug product under varied reconstitution conditions (e.g., degree of shaking), and use of in-line filters.”
The COVID-19 pandemic spurred a lot of innovative drug discovery and development, and this creates new challenges and considerations when it comes to labeling nomenclature. “If there is no USP monograph for the liposome drug product, refer to 21 CFR 299.4, USP General Chapter <1121> Nomenclature, and the USP Nomenclature Guidelines. The liposome drug product nonproprietary name should include terminology to express that the product is a liposome or a pegylated liposome.”
It is possible that nonliposome and liposome drugs can behave differently, though they may contain the same drug substance (active ingredient). This must be taken into consideration, and FDA specifically states: “The Dosage and Administration section should include a statement recommending against substituting the liposome drug product for the nonliposome product or another liposome drug product that contains the same active ingredient unless FDA has determined that the products are therapeutically equivalent.”
In the age of biosimilars, careful attention must be placed on the terminology used to describe drug substitutions. FDA makes this clear in the April 2018 guidance on liposome therapeutics and this draft guidance. The draft states: “FDA recommends avoiding the term interchangeable in this section of labeling because it could create confusion with the same term which is generally used in an interchangeable biosimilarity statement in the Highlights of Prescribing Information of interchangeable biosimilar products.”
Routine Reviews Will Help Ensure Label Accuracy
Due to the increased complexity associated with biological drugs, it is imperative to include all the critical information for the safe and effective use of the drug prior to the Dosage and Administration section. Many of the cell and gene therapies that are being introduced into the clinical pipeline have very specific handling instructions and dosage regimens.
As a reminder, the Dosage and Administration section of the label should be reviewed, at a minimum, annually to be sure that all of the information remains accurate – especially as late-stage trials and commercialization of the product draw closer. Biosimilars will only become more prevalent with time, and it is appropriate that FDA has revamped the 2010 guidance to account for these advances in technology.
About The Author:
 Alexander Aust, M.S., MBA, is the co-founder and owner of Aust Business Solutions, a pharmaceutical consultancy based in Maryville, Tennessee, where he focuses on strategic planning and partnerships, and technical support. The firm provides CMC and project management services, assists with regulatory submissions, and helps to implement new technology. Aust serves as a senior project manager for the Parenteral Drug Association. He earned his MBA from Ball State University and an M.S. degree from Purdue University in molecular and cellular biology.
Alexander Aust, M.S., MBA, is the co-founder and owner of Aust Business Solutions, a pharmaceutical consultancy based in Maryville, Tennessee, where he focuses on strategic planning and partnerships, and technical support. The firm provides CMC and project management services, assists with regulatory submissions, and helps to implement new technology. Aust serves as a senior project manager for the Parenteral Drug Association. He earned his MBA from Ball State University and an M.S. degree from Purdue University in molecular and cellular biology.
References
- FDA Guidance for Industry – Dosage and Administration Section of Labeling for Human Prescription Drug and Biological Products – Content and Format, March 2010.
- FDA Draft Guidance for Industry – Dosage and Administration Section of Labeling for Human Prescription Drug and Biological Products – Content and Format, January 2023.
- FDA Guidance for Industry – Liposome Drug Products – Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation, April 2018.