
Efficient Development Of Oral Drug Products: Selecting A Dissolution Media
By Deanna Mudie Ph.D., Principal Scientist, Lonza

A critical factor formulation experts must understand during the development of an oral solid dosage (OSD) form is that drug and dosage form properties can significantly limit the rate and extent of drug absorption and bioavailability if the OSD is not formulated with these considerations in mind. This lenads to a risk of suboptimal drug delivery in the human body. In vivo dissolution testing is the gold standard for assessing oral bioperformance; however, a key component of this method is the dissolution media you use during testing.
With so many different media available that vary widely in properties and composition, it is important to select one that is tailored to the target population(s) of interest (i.e., fasted humans, fed humans, dogs, etc.) and can capture key gastrointestinal (GI) fluid properties affecting dissolution for your particular drug product.
Assessing Bioperformance In Poorly Soluble Drugs
A solid oral dosage form, like an immediate release tablet, needs to disintegrate into drug particles or granules that are then emptied into the small intestine. These particles must dissolve into solution, which may occur in the stomach and/or small intestine, and also have the potential to precipitate out of solution. Solid particles and dissolved drug then travel down the small intestine and drug in solution is absorbed across the GI membrane into the bloodstream. However, poorly soluble drugs, such as those that fall into the Biopharmaceutical Classification System (BCS) Class II or IV, can be limited by dissolution rate and/or solubility in the GI tract. Therefore, enabled formulations, such as amorphous solid dispersions (ASDs), may be needed to boost their performance.
Physiological, drug substance, and excipient properties can impact in vivo solubility and dissolution rate. Some key drug and excipient properties include intrinsic solubility, acid/base character and lipophilicity, among others, while some key physiological properties include pH, buffer species and concentration, and the presence of bile salts and lipids that can form mixed lipidic structures in the intestine. Factors, such as dissolution rate and in vivo performance, are specific to the drug and the formulation and are dependent on the species of interest and factors that include age, disease state, and whether the subject is fasted or fed. Thus, to develop robust drug products, formulation experts must utilize methods that can help them better understand and predict in vivo performance as a function of these complicated factors. The predominant method used for assessing bioperformance is in vivo dissolution testing. In addition to understanding how to design and implement various testing methods that can accurately identify and help a drug’s performance in the human body, you must also know how to select the most appropriate media for your test.
How To Select The Best Media For Bioperformance Testing Of Your Drug Product
As dissolution testing apparatuses have evolved with our understanding of GI physiology, so too have the dissolution media used during testing. Table 1 below shows the evolution of fluid development to simulate fasted humans, starting with the fluid published by the USP in 1996 and continuing with fluids that became more physiologically relevant and, in some ways, more complex.
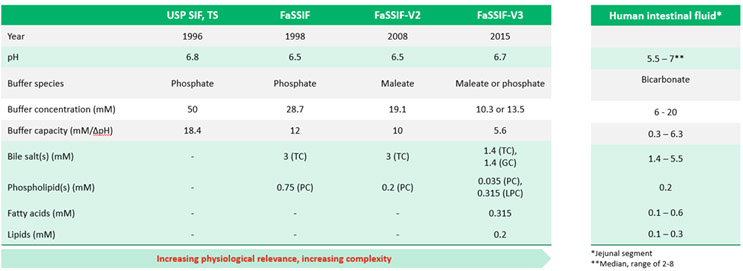
Fluid pH has remained relatively constant within reported median values for human intestinal fluids. Buffer species and concentration have changed, with an effort to bring buffer pKa closer to that of the dominant species in the fastest small intestine, which is bicarbonate, as well as a downward trend toward lower concentration and capacity to be more in line with intestinal fluid. Concentrations of bile salts, lipids, and additional components, which can act to solubilize drug, have evolved to match compositions closer to that of intestinal fluid. Therefore, while some scientists may select the most up-to-date fluid, others rely on earlier versions due to reasons like commercial availability, having a historical data set in a particular fluid, and also practicality. Having so many options can make selection of the most practical, yet biorelevant, media challenging and may lead to questions about whether the chosen media will provide accurate in vivo predictive results.
To solve this challenge, the following methodology aids in selecting practical yet biorelevant media for poorly soluble drugs, focusing on fasted state humans. This methodology is based on the interplay between GI fluid properties and drug or excipient properties, taking the following three key interactions into account.1
- pH, acid/base character, and pKa
- buffer species and concentration, acid/base character, pKa, and intrinsic solubility
- bile salts and lipids in the fluid and lipophilicity of the drug
Investigating these concepts provides a basis for choosing pH, buffer species and concentration, and inclusion of bile salts and lipids with the goal of simplifying the media as much as possible while allowing us to achieve physiologically meaningful results. Table 2 is an overall summary of recommendations for selection of the drug/excipient properties listed above.
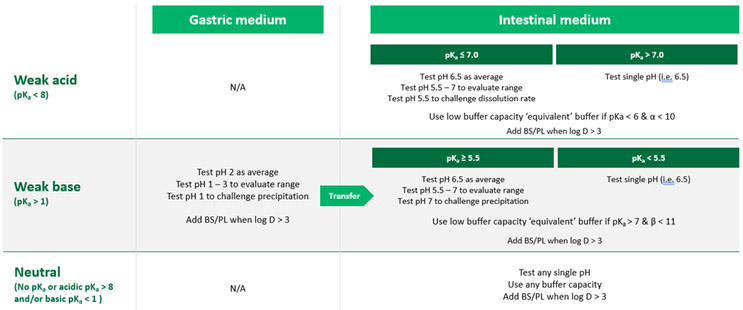
The following sections detail the bases for these recommendations.
Selection Of Medium pH
In fasted humans, the average pH ranges from about 1 to 3 in the stomach, and it increases to about 5.5 to 7 in the upper small intestine. GI fluid pH can impact solubility (shown as Cs in the equation below) and, therefore, the dissolution rate (represented as dM over dt in the same equation). The acid/base character and pKa of the drug affects the solubility of the drug or excipient at a given pH by impacting the extent of ionization, with a higher degree of ionization resulting in higher solubility.
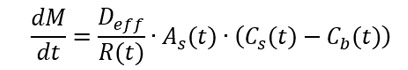
The three graphs in Figure 2 show the relative solubilities of compounds versus fluid pH, according to the Henderson-Hasselbalch equation, where there is an increase in solubility compared to the intrinsic solubility of the molecule as a result of ionization.
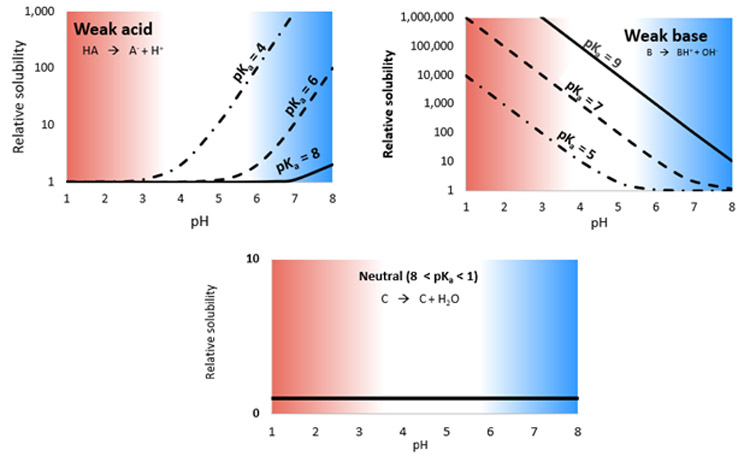
The top left of Figure 2 shows the relative solubilities of example weak acids with pKa values ranging from 4 to 8. Solubility is low and equal to the intrinsic solubility at low pH but increases substantially as pH increases and the drug ionizes. Solubility is also dependent on the drug pKa. The opposite is true for weak bases (shown on the upper right graph), which tend to ionize at low pH and, therefore, have higher solubility, but precipitate at increasing pH as the solubility decreases. Since neutral compounds do not ionize in the physiological pH range, relative solubility is equal to the intrinsic solubility and is constant along the pH range. Thus, weak acids have increased solubility and, as a result, an increased dissolution rate at higher intestinal pH, whereas weak bases have increased solubility and, therefore, increased dissolution rate at lower gastric pH. Neutral compounds show no pH dependence.
Selection Of Buffer Species And Concentration
When considering buffer selection, it is important to consider the interplay between the buffer species and the acid/base character, pKa, and intrinsic solubility of the drug or the excipient. While 0.5 to 20 millimolar (mM) hydrochloric acid is the dominant component in the stomach, bicarbonate is the dominant buffer species in the fasted small intestine at concentrations in the range of about 6 to 20 mM, resulting in relatively low buffer capacities.1 Despite being the physiological buffer, bicarbonate buffer is rarely used for in vivo dissolution testing since it is more difficult and time consuming to prepare than commonly used buffers, such as phosphate. However, using non physiological buffers, like high buffer concentration phosphate, may not be biopredictive since buffer species and concentration can greatly impact dissolution rate for some weak acids and bases.
Figure 3 shows flux, which is the surface-area normalized dissolution rate versus the buffer concentration for the weak-base drug Haloperidol, dissolving in a commonly used phosphate buffer (represented by the square data points); and in a bicarbonate buffer (represented by the circle data points).
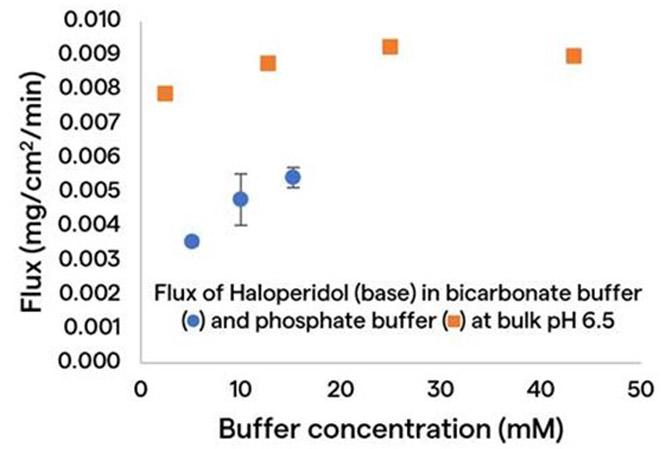
As shown in Figure 3, dissolution rate increases with an overall upward trend in buffer concentration in both buffers, and at a given buffer concentration, the dissolution rate of the same drug in the bicarbonate buffer is much slower than it is in the phosphate buffer. These results make it evident that, in order to project a physiologically relevant dissolution rate, one may need to either use a bicarbonate buffer or select a surrogate buffer that can match performance in bicarbonate.
The mechanism for these buffer effects is through the pH at the surface of the dissolving drug particle, as shown on the left in Figure 4.

As previously described, pH impacts solubility (represented by CS in the figure above) of weak acids and bases, which affects the dissolution driving force. It is important to differentiate between pH at the solid particle surface (pHS) from the pH in the bulk solution (pHb) since pHS is what impacts CS, i.e., the solubility at the surface. The reason this distinction should be made is because certain weak acids and bases can change the surface pH when they dissolve, making the surface pH different from the bulk pH when the buffer capacity at the surface is not high enough to withstand the pH change. Therefore, solubility and dissolution rate are lower compared to a case where the buffering capacity was high enough to withstand a potential pH change caused by dissolution of a weak acid or base. This results in dissolution that is sensitive to buffer species and concentration.
Not all weak acids and bases change the surface pH as they dissolve, though, so not all are sensitive to the selected buffer species and concentration. When a weak acid dissolves (shown on the upper right in Figure 4), the propensity to lower the surface pH increases as the pKa decreases and the intrinsic solubility (represented by S0) increases. However, when a weak base dissolves, the propensity to raise the surface pH increases as pKa increases and intrinsic solubility increases. In both cases, this can reduce the dissolution rate.
Figure 5 shows that the factors of pKa and intrinsic solubility can be combined to determine when buffer effects are important, using weak acids as an example.
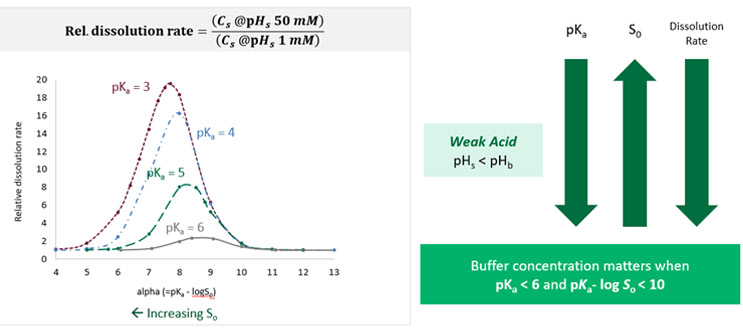
The plot on the left shows the calculated relative dissolution rate in a 50mM concentration of phosphate buffer compared to a 1mM concentration of phosphate buffer for drugs with different pKas as a function of the parameter alpha, which is an expression that takes into account drug pKa and intrinsic solubility. In this example, the relative dissolution rate is sensitive both to pKa and to the parameter alpha. For a drug with a pKa of 3 (shown in the maroon color data points), relative dissolution rate is around 20, meaning there is a 20-fold difference in dissolution rate between the two buffer concentrations. This occurs at an alpha value of around 7.5.
However, when alpha is > 10, we see no difference in dissolution rate, meaning the drug is not sensitive to the buffer concentration in this range of alpha. When looking at a drug with a pKa of 6 (shown in the gray data points), there is only a maximum twofold difference in dissolution rate at an alpha value of around 8.5. Again, though, there is no difference above an alpha value of 10. This analysis can be used as a paper exercise for determining when buffer species and concentration will be important. For weak acids, the buffer species and concentration are predicted to be important when pKa is < 6 and alpha is < 10.
Selection Of Bile Salts And Lipids
When deciding whether to include bile salts and lipids in the media, the interplay between the bile salts and lipids in vivo and the lipophilicity of the drug or the excipient should be considered. In fasted humans, there is a residual amount of bile salts and lipids in the stomach but a more substantial amount ranging from about 1.4 to 5.9 bile salts mM and about 0.2 mM phospholipids in the small intestine. These main components, together with fatty acids and cholesterol, can interact and form mixed lipidic structures of micelles and vesicles. Drug in solution can then bind to these structures, which further solubilizes the drug above the solubility that would be achieved in the absence of these structures. Figure 6 shows the apparent solubility increase in the presence of these structures as a function of lipophilicity for about 80 different compounds (represented by the calculated drug distribution coefficient, log D, at pH 6.5).
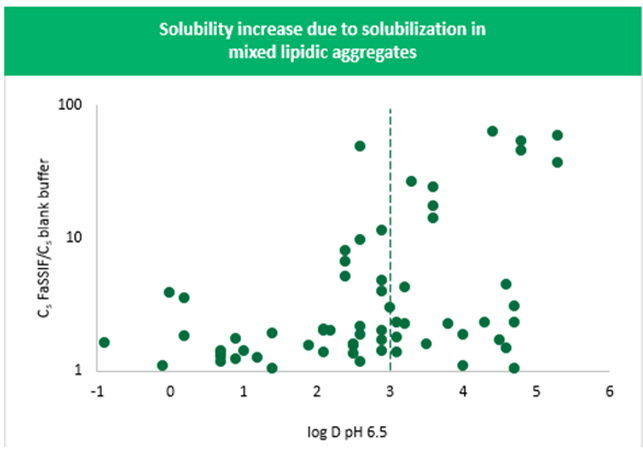
Specifically, this plot shows the solubility in FaSSIF (contains 3 mM of bile salts and 0.75 mM phospholipids) divided by the solubility in a blank medium (does not contain these bile salts or phospholipids) versus drug log D at pH 6.5. The plot indicates a general trend toward an increase in solubilization with an increase in log D at pH 6.5. The solubility increase exceeds a value of 10 when log D is > 3. While the true increase in solubility for a given compound can most accurately be determined by measuring the partition coefficient in the micelles or vesicles themselves, using predicted log D as a surrogate provides a convenient indicator without the need for experimentation.
Solubilization in these structures can also impact dissolution rate. In addition to impacting Cs and Cb in the dissolution equation, it is also important to consider the impact on the apparent diffusion coefficient (represented by Deff in Equation 1). The apparent diffusion coefficient describes the rate of diffusion of a drug from the particle surface, with a higher Deff resulting in a faster dissolution rate. When both free unbound drug and drug bound to micelles or vesicles are present, the Deff value can be calculated as a weighted average of the unbound drug diffusion coefficient times the fraction of unbound drug plus the micelle or vesicle diffusion coefficient times the fraction of bound drug (Du × fu + Dm × fm).
Although the apparent solubility may increase when adding bile salts and lipids to the medium, the dissolution rate may not actually increase. This is due to the competing impact of apparent diffusion coefficient and apparent solubility on the dissolution rate. Effective diffusion coefficient is relatively high for free drug (Figure 7) because the molecule is smaller than that of the micelle or vesicle, but solubility may be relatively low. However, in the case of drug bound to micelles or vesicles, diffusion coefficient decreases due to the increase in size of the micelle or vesicle compared to the free drug, whereas apparent drug solubility increases due to solubilization in the micelle or vesicle.

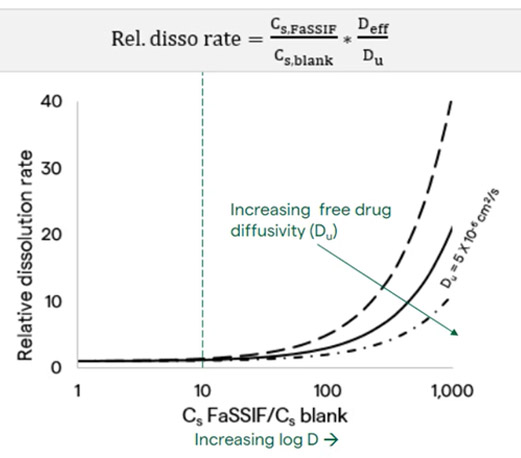
A tenfold increase in solubility results in a minor increase in dissolution rate, and it is not until there is a 100-fold increase in solubility that we see up to a fivefold increase in dissolution rate. This continues to increase as the difference in the solubility goes up. Because a larger than tenfold increase in solubility is typically only seen for drugs with a log D > 3 at pH 6.5 (Figure 6), it can be assumed that when drug log D < 3 at pH 6.5, there would be no appreciable change in dissolution rate.
While the above analyses are based upon calculations, the media selection recommendations presented in Table 2 were confirmed by testing model compounds, and by finding example reports in the literature, as described in the open access, peer-reviewed publication, Selection of In Vivo Predictive Dissolution Media Using Drug Substance and Physiological Properties.1 In addition, application of this selection methodology to media selection for an amorphous solid dispersion formulation can be found in the publication, Use of biorelevant dissolution media to accelerate development and increase robustness of oral drug products.7
Summary
Selecting a dissolution media that can rank or test formulation robustness is important for the efficient development of oral drug products. Knowledge about key physiological and drug formulation properties can be combined to streamline media selection, enabling Right First Time development while minimizing fluid complexity. This selection approach can ultimately reduce costs and increase development speed of medicines.
- Mudie DM, Samiei N, Marshall DJ, Amidon GE, Bergström CAS. Selection of In Vivo Predictive Dissolution Media Using Drug Substance and Physiological Properties. AAPS J. 22, 2 (January 2020):34. doi: 10.1208/s12248-020-0417-8.
- Stewart AM, Grass ME, Mudie DM, Morgen MM, Friesen DT, Vodak DT. Development of a Biorelevant, Material-Sparing Membrane Flux Test for Rapid Screening of Bioavailability-Enhancing Drug Product Formulations. Mol Pharm. 5;14(June 2017): 2032-2046.
- Fuchs A, Dressman JB. Composition and physicochemical properties of fasted-state human duodenal and jejunal fluid: a critical evaluation of the available data. J Pharm Sci. 2014 Nov;103(11):3398-3411
- Wang Y & Brasseur JG. Mol Pharm. 2012 May 7;9(5):1052-66
- Bergstrom CAS et al. Eur J Pharm Sci. 2014
- Tsume, Y. et al. Eur J Pharm Sci. 2014., Koenigsknecht et al. Mol Pharm. 2017., Bergstrom CAS et al. Eur J Pharm Sci. 2014
- Mudie, Deanna. (July 28, 2021). Use of biorelevant dissolution media to accelerate development and increase robustness of oral drug products. European Pharmaceutical Manufacturer. https://www.epmmagazine.com/pharmaceutical-industry-insights/pharmaceutical-manufacturing-insights/use-of-biorelevant-dissolution-media-to-accelerate-developme/