
EMA's Revised Format For Risk Management Plans – What You Need To Know
By Anusha Akula, Freyr Solutions

Pharmacovigilance (PV) has been undergoing continuous transformation over the years with respect to evolving processes, technology, legislation, and guidelines to ensure enhanced patient safety and improved monitoring of the safety profile of medicinal products. Since the execution of the International Conference on Harmonization (ICH) E2E Guidelines in 2004,1 proactive risk management strategies have significantly progressed at a global level in the PV domain. Over the years, the global perspective diverged into a U.S. FDA concept and a European concept governed by the European Medical Agency (EMA). The U.S. risk management strategy started in 2005 with the Risk Minimization Action Plans (RiskMAPs).2 The current format of the Risk Evaluation and Mitigation Strategies (REMS), which replaced the RiskMAPs, has been enforced since 20073 through the Food and Drug Administration Amendments Act (FDAAA).
The European risk management strategy was first implemented in a Guideline On Risk Management Systems For Medicinal Products For Human Use in 2005.4 It was then developed as a template for the EU Risk Management Plan (RMP) in Volume 9A of The Rules Governing Medicinal Products in the European Union in 2006.5 Later, it was described and updated in the Good Pharmacovigilance Practices (GVP) modules as ‘Guidance on the format of the risk-management plan in the European Union – in integrated format’.6 Revision 1 of the guidance was released in April 2014,7 and the latest update, Revision 2 of the guideline8 and guidance on the format of RMP,9 was released in March 2017. Revision 2 is effective from March 31, 2017, and the Revision 1 format was accepted only until March 31, 2018. It has been observed that most of the rest of the world’s health authorities maintain their own format for RMPs, which is quite like the earlier version of the EMA RMP format.
Generally, the strategy of both concepts for risk management (REMS and RMP) is to manage and prevent the known or potential serious risks associated with a medicinal product to ensure that the benefits outweigh risks. Each REMS is designed to address a specific serious safety concern, including information communicated to and/or required activities to be undertaken by healthcare providers, pharmacists, and patients (elements to assure safe use). REMS is generally required by the FDA, depending on the size of population likely to use the medicine, the seriousness of the disease, expected duration of treatment, expected benefits of the drug, and seriousness of known or potential risks. RMP consists of a medicine's complete safety profile; how its risks will be prevented or minimized; plans for further studies and other PV activities to gain more knowledge about the safety and efficacy of the medicine; and effectiveness of risk-minimization measures. The EMA requires submission of the RMP at the time of application for a marketing authorization of a medicine in the EU and additionally as required by a national competent authority whenever information affecting the benefit-risk balance of a medicine is available. RMPs are continually modified and updated throughout the product’s life cycle as and when new or significant information is available.
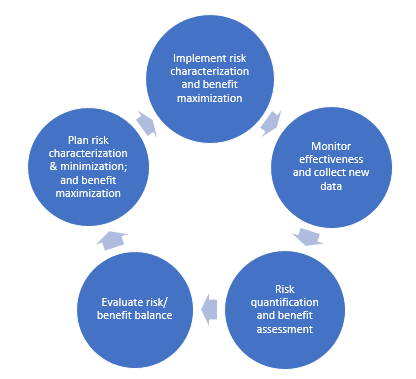
Figure 1: Typical presentation of a risk management cycle
What Has Changed In Revision 2?
The revised format for RMP sets a new milestone in a progressive approach to risk management. The new RMP template is a straightforward and well-structured document that can be used by RMP experts, and the concepts behind risk management have been justified to better reflect the stages of the life span of a medicinal product. Revision 2 of GVP module V addresses most of the areas identified for improvement based on experience and feedback to EMA and other stakeholders.
Major revisions include:
- Further clarification on what the RMP should focus on with respect to an important identified risk, important potential risk, and missing information
- Guidance on the expected changes in the RMP during a product’s life cycle
- Removal of duplication within RMPs and other submission documents
- Updated minimum requirements for various initial marketing authorization applications (MAAs)
- An amended RMP template
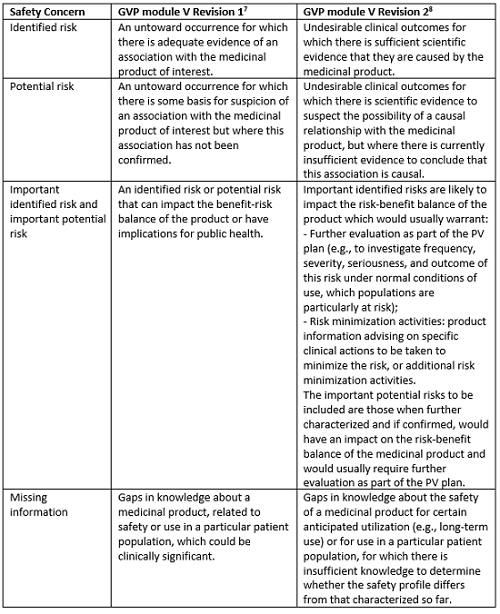
Redefined Safety Concerns
The major change in the revised guidance on RMP has been with respect to safety concerns, which ensures RMP will be more risk-proportionate in terms of consideration criteria and characterization of important identified risks, important potential risks, and the missing information. The safety concerns are now more precisely defined, helpful in understanding what is relevant for inclusion in safety specification (Part II) of the RMP, how the important risks are characterized, and how the safety concerns evolve through the life span of the medicinal product.
Table 1: Differences in Definitions of Safety Concerns in GVP Module V Revision 1 and Revision 2
Evolution Of Safety Concerns Through The Product’s Life Cycle
The information required to be provided in the safety specification of the RMP varies depending on the stage of the product’s life cycle and the need for post-authorization data. It is well known that a full RMP should be submitted for initial MAAs, whereas for products with an established safety profile and post-marketing knowledge (e.g., generic drugs, fixed-drug combinations), most modules of the safety specification are not required. Similarly, risk-proportionate RMP in a true sense implies that knowledge regarding a medicinal product’s safety profile is expected to increase and safety concerns are expected to evolve through the product’s life cycle. The GVP module V Revision 2 provides guidance on the removal of safety concerns from the existing RMP post-authorization. This encourages marketing authorization holders (MAHs) to critically revise the list of safety concerns, PV activities, and risk minimization measures in the post-marketing phase. The product’s safety profile will thus change, confirming or refuting a causal association with the medicinal product. In addition, PV activities and risk minimization measures may also change over time. Duplication of the information on identified and potential risks already covered in the safety sections of the dossier, including signal evaluation, periodic benefit-risk evaluation, or safety variations procedures, is avoided. The module SVII is now confined to “New safety concerns and reclassification with a submission of an updated RMP.”
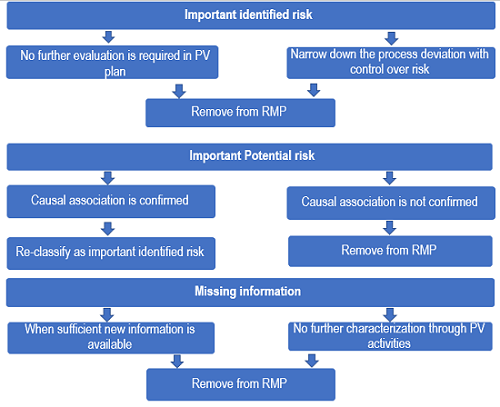
Figure 2: Evolution of safety concerns over time through the product’s life cycle
Other Changes In Safety Specification
The revised guideline states that the RMP should provide summary information of the patients studied in clinical trials in an appropriate format (e.g., tables/graphs) in the clinical trial exposure section (module SIII) of the initial RMP or when there is a major update due to new exposure data from clinical studies (e.g., in a new indication). In the case of absence of new significant exposure data, this section is not required to be updated. The excluded populations from the clinical trial development program should be included as missing information in module SIV, “Populations not studied in clinical trials,” only for approved and proposed indications and if the use in such populations might be associated with any risks of clinical significance. It has now been made clear that the information on post-authorization experience of the product in other regions outside the EU or from other authorized products with the same active substance from the same MAH should also be discussed in module SV, “Post-authorisation experience.”
Plans For Post-Authorization Efficacy Studies (PAES)
The scope of the modules for PV plan and post-authorization efficacy studies is now more restricted with clear understanding. The revised format of RMP is confined to a list of post-authorization efficacy studies imposed as conditions to the marketing authorization or when included as specific obligations in the context of a conditional marketing authorization or a marketing authorization under exceptional circumstances.
Summary Of Changes To The Risk Management Plan Over Time
A new annex 8 has been added to the revised format, which lists all the significant changes to the RMP in chronological order, with the date and version number of the RMPs prepared. This should mention if any safety concerns were added, removed, or reclassified; whether any studies were added or removed from the PV plan; and whether any risk minimization activities were modified in the risk minimization plan.
Changes In The Template
The name and signature of the qualified person for pharmacovigilance (QPPV) should be presented on the title page to ensure that the RMP has been reviewed and approved by the MAH/applicant’s QPPV and that the electronic signature is documented.
The revised guidance text is streamlined by removing duplications within the RMP modules or in other safety reports, including the periodic safety update report (PSUR) or elsewhere in dossier submission. The update focuses on conciseness in sections that often contain duplicate information, including the summary table of safety concerns, epidemiology, populations not studied in clinical trials, summary of treatment results, etc. The summary of minimum RMP requirements for different initial MAAs (full MA application, generic product, fixed combination product, biosimilars, etc.) allows the stakeholders to clearly understand the information to be included in various sections of the RMP.
Other changes in Revision 2 are small and simple. For example, the Part I Product Overview of the RMP does not require the date and country of first worldwide authorization and launch to be recorded.
Most of the sections are largely unchanged from the original document, but there are some significant differences. The mapping between RMP modules and information in the electronic common technical document (eCTD) remains the same in the revision.
Challenges With The Revision
The organizations working on RMPs should update their standard operating procedures and work instructions with new definitions of terminology of safety concerns. With the revision of the RMP format, there also exists a major challenge in terms of time, data integrity, and regulatory compliance with the transfer of content from the existing template to the new template when an RMP update is required. Converting an RMP from the Revision 1 format to Revision 2 is not a simple job of condensing or transcribing the content. There have been issues observed while making the changes in a “track change” version due to the addition and removal of sections in the new format. However, with the streamlined process and the expertise of the authors involved, it is not an impossible task. The scope of the revised RMP format is substantial. A question always remains as to whether MAHs will be able to justify the available evidence enough for a critical review, and whether the assessors from the agency will agree to the proposed changes.
Moving Forward
Since April 1, 2018, EMA requires that MAHs submit RMPs in the Revision 2 format for all initial MAAs, D91, and D121 responses and for RMP updates. Health authorities in the rest-of-the-world (ROW) countries requiring the EMA RMP Revision 1 format have also adapted to the revised format. The authorities of other countries requiring their own specific templates, like Mexico, Chile, etc., have been continuing with the same for RMP submissions.
In the near future, we can hope and expect the health authorities of all the ROW countries to adapt to the EMA Revision 2 format of the RMP to ensure a consistent global risk management system for medicinal products, with a prospect of better patient safety.
Conclusion
Revision 2 of GVP module V results in simpler RMPs. The safety concerns are well redefined to clarify relevant information to be incorporated in the RMP of a medicinal product, leading to RMPs that are not loaded with information on risks already covered in other documents. Indeed, the focus is on identifying or characterizing the safety profile of the medicinal product, proposing measures to prevent or minimize the risks, and including an assessment of the effectiveness of the proposed measures. The revised RMP format provides a clear, focused, and scientifically justified vision for risk management, as well as saving companies time through a more concise, less repetitive approach.
References:
- Pharmacovigilance Planning E2E. ICH Harmonized Tripartite Guideline. 2004 Nov 18. [cited 2018]. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2E/Step4/E2E_Guideline.pdf.
- Guidance for Industry – Development and Use of Risk Minimization Action Plans. U.S. FDA. 2005 Mar. [cited 2018 Aug 10]. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceRegulatoryinformation/guidances/ucm071616.pdf.
- Risk Evaluation and Mitigation Strategies: Modifications and Revisions Guidance for Industry. U.S. FDA. 2015 Apr. [cited 2018 Aug 10]. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceRegulatoryinformation/guidances/ucm441226.pdf.
- Guideline on Risk Management Systems for Medicinal Products for Human Use, EMA/838713/2011. CHMP. EMA. 2005 Nov 14 [cited 2018 Aug 10]. Available from: https://www.emwa.org/Documents/Freelancer/riskmanagement/rmp%20guidelines.pdf.
- Annex C: Template for EU Risk Management Plan, EMA/838713/2011. EMA. 2006 Sep 27 [cited 2018 Aug 10]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Template_or_form/2009/10/WC500006317.pdf.
- Guideline on good pharmacovigilance practices Module V – Risk management systems, EMA/838713/2011. EMA and HMA. 2012 Feb 20 [cited 2018 Aug 10]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/02/WC500123208.pdf.
- Guideline on good pharmacovigilance practices (GVP) Module V – Risk management systems (Rev 1), EMA/838713/2011 Rev 1*. EMA and HMA. 2014 Apr 15 [cited 2018 Aug 10]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/03/WC500224570.pdf.
- Guideline on good pharmacovigilance practices Module V – Risk management systems (Rev 2), EMA/838713/2011 Rev 2. EMA and HMA. 2017 Mar 28 [cited 2018 Aug 10]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129134.pdf.
- Guidance on the format of the risk management plan in the EU – in integrated format, EMA/PRAC/613102/2015 Rev.2. accompanying GVP Module V Rev. 2. EMA. 2017 Mar 30 [cited 2018 Aug 10]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2017/03/WC500224771.pdf.
About The Author:
 Anusha Akula is a professional medical writer with over five years of experience in pharmacovigilance and clinical development domains. In addition to having postgraduate degree in pharmaceutical sciences, she is also a certified professional in pharmacovigilance and SAS. She has worked for innovator and generic pharmaceutical companies and gained extensive experience in writing and quality review of various regulatory documents for U.S., EU, and ROW submissions. She currently works as a senior medical writer at Freyr Solutions, India.
Anusha Akula is a professional medical writer with over five years of experience in pharmacovigilance and clinical development domains. In addition to having postgraduate degree in pharmaceutical sciences, she is also a certified professional in pharmacovigilance and SAS. She has worked for innovator and generic pharmaceutical companies and gained extensive experience in writing and quality review of various regulatory documents for U.S., EU, and ROW submissions. She currently works as a senior medical writer at Freyr Solutions, India.