
FDA Explains How To Respond To Form 483 Observations In New Draft Guidance
By Peter H. Calcott, Ph.D., FRSC, president and CEO, Calcott Consulting LLC

In March 2026, the FDA issued a new draft guidance titled Responding to FDA Form 483 Observations at the Conclusion of a Drug CGMP Inspection Guidance for Industry.1 The FDA requested comments via docket number FDA-2025-D-1504 and comments were due by May 8.
For those experienced and successful at responding to Form 483 observations from the FDA, there might be an opinion that this guidance contains nothing new. But that would be an oversimplification. There are several elements in this document that eloquently spell out the agency’s expectations, which I believe are often lacking in many responses from companies. This is the first time, at least from my perspective, that the FDA has articulated these expectations. As you walk through this document, these elements are described with the logic explained. I will point out those elements as I describe the guidance content and scope.
The introduction does not reference any previous source of their expectations; it simply quotes various GMP principles. They articulate that the issuance of a Form 483 is to describe to the company observations that were made in an inspection that potentially could mean that the operations examined are not meeting cGMP. The form is an invitation for dialogue between the agency and company and for the company to defend or explain the observations and actions seen and their position. This allows the company to explain their systems and describe how they are operating under GMP or to propose other ways to satisfy the observations. This discussion should include a detailed discussion of the circumstances that led to the observations and a deep discussion on the scope and ramifications of the observations. The response can address the written observations as well as the oral observations delivered in the closing meeting (and included in the Establishment Inspection Report).
This actually sounds like it is possible to neutralize the observations in the response, but bear in mind you have already spent perhaps five days trying to explain during the actual inspection. In practice, while this can happen (it has happened to me several times), the vast majority of observations will need some form of correction. The agency points out that this needs to be described well in the response.
Your response to this set of observations will pave the road to what will happen later, although it should be noted that some Form 483 observations are such that a warning letter will ensue even with the best crafted response. But even in those cases, a well-crafted response is part of the well managed preparatory response to the subsequent warning letter.
Submitting A Response To The FDA
Section III of the guidance describes what needs to be in the response and its content. The response document needs to be self-contained so a reviewer does not have to open other documents. The FDA describes the important elements, which I’ve compiled/paraphrased into Table 1.
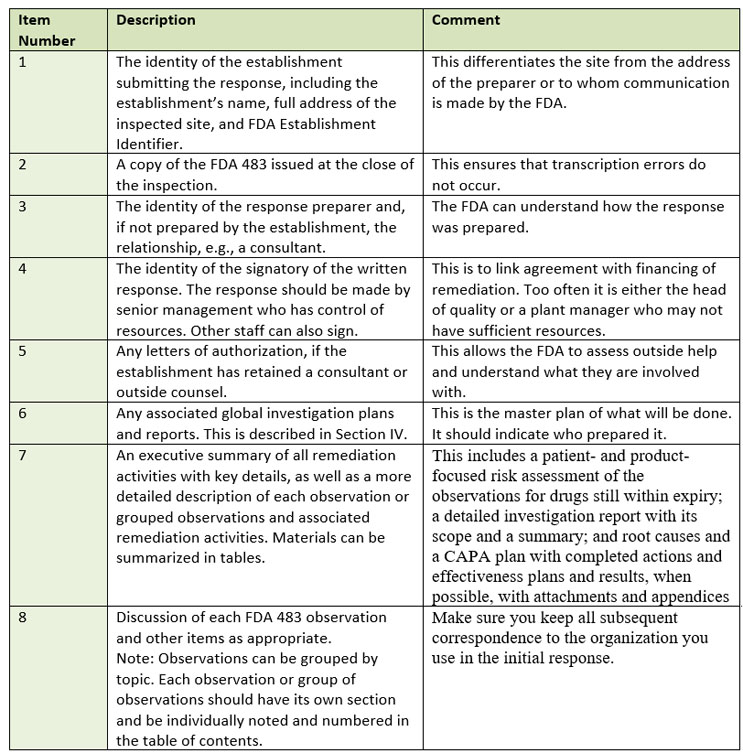
Table 1: What must be included in the response?
Several elements do stand out. Firstly, the FDA advises that the signer of the response should be senior management with access to adequate resources. Clearly this is to assure that the fiscal impact of remediation is considered and approved. During most of my career, it was customary for the head of quality at the site to sign the response and/or sometimes the plant manager. Secondly, the guidance is clear that if you use a consultant, they want to know who it is, their qualifications, and what they are contributing. Thirdly, while all observations must be addressed, they can be grouped into like observations. This might result in two observations being grouped into one response or an observation split and grouped with similar observations in a second citation. In other words, group according to logic.
With a response being due in 15 working days, it is often the case that not all items can be satisfactorily addressed immediately, so the response will include a detailed plan of what will be done, along with a timeline. I have always advised clients not to be overly optimistic in the timeline but rather recognize things will often not proceed as quickly as planned. So, give yourself time, but deliver on time. Describe when follow-ups will occur. I espouse under-promising and overdelivering. The FDA even advises where to send the response via email or via other electronic gateways (for big files) or how to contact them if delivery is an issue.
Addressing Form 483 Observations
Section IV contains many of the very useful recommendations contained in the guidance. As described before, grouping observations or sub-observations by commonality makes sense, especially if the corrective actions and preventative actions (CAPA) are similar. If you start with the groupings, make sure to retain the same groups for follow-up or subsequent submissions. At this stage it is wise to assess the relative severity of each observation with respect to patient safety and efficacy and whether the observation is systemic in nature. This will allow you to focus on the more critical observations. In addition, examine the results of your audit program and previous inspections to see if the observations are new or are already known. Assess why previous CAPAs failed or are not completed yet if it is a known issue.
To quote the FDA: “FDA observations are not an exhaustive list of all deficiencies that could be present at an establishment.” So, your response should look and probe deeper than the actual observation. Think holistically in your analysis. Go beyond the cited observations and see if there are more that the agency did not identify (other instances and other locations). With every observation ask why the system allowed this observation to occur. This is one of the elements I have found missing in many responses by companies.
Sometimes it is wise to employ a consultant when either your organization is not highly experienced at dealing with inspections or you sense the observations are very serious. This consultant can add expertise but also convince the agency you have taken the observations seriously. A well-crafted curriculum vitae with a contract scope of work add to the credibility of the consultant and their engagement with the company.
The first stage of action is understanding and assessing the observations. There should be a link between observation and severity so you can prioritize. You should also determine how systemic the issue is or whether it is a one-off. The FDA suggests several systems to examine to gather information, including complaints, verbal FDA observations, interviews with employees, document and record review, and regulations, guidances, and FDA presentations. This activity sets the stage for the remediation.
Management plays a central role in surviving an inspection. You need management support for all remediation actions, so they must be in the loop and engaged at all stages. There is nothing worse than promising something in a response, but management fails to deliver the resources for correction.
The next stage after the initial analysis is to develop an action plan. This plan should examine each grouping of observations and determine the impact of the observation on a number of elements. Firstly, determine whether the observation is limited to just the product involved in the observation. Some observations point to broader issues that could impact multiple products in your portfolio or even all operations. Once the scope of impact is determined, define what product on the market is still within expiry. A risk assessment on the impact of this observation on the product is critical. At the worst, a recall might be considered as part of the actions. A clear defense of product on the market must be considered at the bare minimum. In my 20 years consulting, I have had several clients that have initially not considered this element in their response.
For each observation, you must perform a root cause analysis asking why the events leading to the observation occurred. The root cause may be a single failure, but often it is several elements that have failed or contributed. So do not stop on the first one discovered. If human error is pinpointed and retraining operators is considered, I point you to the FDA’s Quality Metrics Program for advice.2 Uncovering the root cause is but the first step in the root cause analysis. The next question the FDA wants answered is: Why was the quality unit unaware of this potential deficiency? What specific quality system failed to detect or caused this? In my experience this is an area that companies miss and do not consider well.
I’ve created Figure 1 to illustrate a hypothetical observation (an operator was not trained on a specific task). Obviously, the operator needs to be trained and that is one correction. But you must ask several further questions (the ones in the figure are some questions that should be posed and not a complete list). Those questions could lead you to questioning several quality systems in this case, including linkage of SOP approval and training requirements and the whole structure of the training program and scheduling, to name two elements. But the root cause goes further: you need to question the audit program’s failure to identify this issue. So, if a simple retraining of operators CAPA has become a comprehensive remedial program of several quality systems. The net has been cast far from the original observation. Of course, other quality systems might be involved and failed in your observations, such as change control or investigations, to name but two.
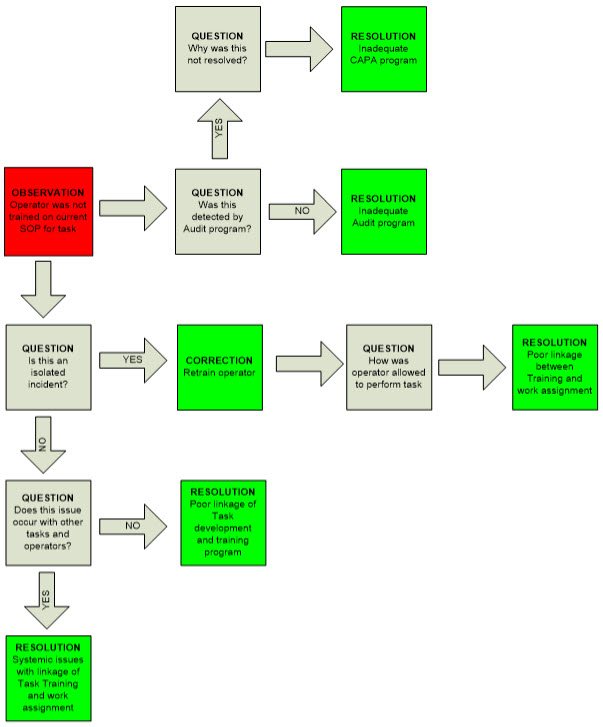
Figure 1: Potential Decision-Making Pathway for a Simple Observation (A hypothetical case to illustrate the principles).
Red = observation
Gray = question
Green = resolution
It’s only after a detailed action plan has been implemented with a thorough root cause analysis performed that CAPA plans can be assembled. As indicated before, if you find retraining operators as your CAPA, you need to recheck that conclusion by casting a wider net. Personally, I also go back to look at whether rewriting the SOP is called for as well. That does not mean that retraining operators cannot be good CAPAs; they are just not for everything. Although the FDA does not stress it, the difference between corrective action and preventive action should be clear. One corrects the problem at hand, while the other prevents it from ever happening. As part of the CAPA plan, describe how you are going to measure effectiveness. Again, based on my experience this is often a weakness in company responses.
After implementing the CAPAs, track them to completion. The FDA stresses that effectiveness must be checked for all CAPAs. The FDA further indicates that the results of the effectiveness checks should be reported to them as part of the submission. After the initial response within 15 days, which includes the overall plan and timeline, interim reports are often submitted as blocks of deliverables are completed. Pay particular attention to the timeline you proposed to the FDA, keep to it, and if it seems like a deliverable is tracking late, do not wait until after the date to inform the FDA. The agency recognizes things go differently to plan and, in my opinion, understands it.
The FDA’s closing section describes mechanisms for disagreement resolution, including the FDA ombudsman. In all cases, present your case with the evidence.
Conclusion
The draft guidance contains several specific takeaways often missed by companies responding to inspection observations:
- Ensure engagement of senior management. They control the purse strings.
- If you use a consultant, make sure it is clear what they are doing and that they are qualified.
- The root cause analysis goes further than the observation and questions what quality system allowed this to happen. That is the true culprit in the game.
References:
- Responding to FDA Form 483 Observations at the Conclusion of a Drug CGMP Inspection, March 2026 Inspection; https://www.fda.gov/media/191427/download.
- Quality Metrics Technical Conformance Guide Technical Specifications Document; July 28, 2015; https://www.fda.gov/media/98939/download
About The Author:
 Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.
Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.