
FDA Seeks Comment On ICH Q9(R1) Quality Risk Management
By Mark Durivage, Quality Systems Compliance LLC
 On June 16, 2022, the FDA’s Center for Drug Evaluation and Research (CDER) released for public comment Q9(R1) Quality Risk Management Guidance for Industry. The International Council for Harmonisation (ICH) of Technical Requirements for Pharmaceuticals for Human Use has undertaken the revision of Q9 Quality Risk Management to ensure that safe, effective, and high-quality medicines are developed, registered, and maintained in the most resource-efficient manner worldwide. The FDA, a founding member of the ICH, plays a major role in the development of each of the ICH guidelines, which FDA then adopts and issues as guidance to industry.
On June 16, 2022, the FDA’s Center for Drug Evaluation and Research (CDER) released for public comment Q9(R1) Quality Risk Management Guidance for Industry. The International Council for Harmonisation (ICH) of Technical Requirements for Pharmaceuticals for Human Use has undertaken the revision of Q9 Quality Risk Management to ensure that safe, effective, and high-quality medicines are developed, registered, and maintained in the most resource-efficient manner worldwide. The FDA, a founding member of the ICH, plays a major role in the development of each of the ICH guidelines, which FDA then adopts and issues as guidance to industry.
The purpose of the guidance is to enhance the application of effective quality risk management principles by industry and regulators and provide a systematic approach to quality risk management for improved, informed, and timely decisions. The FDA recognizes quality risk management is a valuable component of an effective pharmaceutical quality management system to ensure drugs are safe and effective.
Q9(R1) Quality Risk Management provides principles and tools for quality risk management that can be applied throughout the product life cycle of drug substances, drug products, and biological and biotechnological products, including the development, manufacturing, distribution, inspection, and regulatory submission and review processes.
Q9(R1) Quality Risk Management Step 1, sign-off on the consensus draft technical document, was completed in October 2021. It was signed off in November 2021, as a Step 2 document to be issued by the ICH Regulatory Members for public consultation. This document was developed based on a Concept Paper approved November 2020 and a Business Plan approved October 2020. ICH is anticipating finalization as a Step 4 document for implementation by local regional regulatory systems in September 2022.
Principles Of Quality Risk Management
ISO/IEC Guide 51 defines risk as the combination of the probability of occurrence of harm (damage to health, including the damage that can occur from loss of product quality or availability) and the severity (a measure of the possible consequences of a hazard [potential source of harm]) of that harm.
Quality risk management is composed of two fundamental principles. First, evaluation of the risks to quality should be based on scientific knowledge and ultimately link to the protection of the patient. Second, the quality risk management process (effort, formality, and documentation) should be proportionate to the level of risk.
Quality risk management is a systematic process for the assessment, control, review, and communication of risks to drug product quality throughout the drug product’s life cycle. Q9(R1) Quality Risk Management provides a quality risk management model as shown in Figure 1. When risks are determined to be unacceptable due to risk to the patient and statutory or regulatory requirements, the risk should be reassessed. Other risk models may be used but should generally include the same elements as provided in the model shown in Figure 1.
Quality risk management activities are best undertaken by an interdisciplinary team representative of the business activities involved, which may include quality, regulatory affairs, operations, production, sales, marketing, research and development, engineering, maintenance, supply chain, warehousing, distribution, customer service, clinical affairs, and post-market surveillance. Interdisciplinary teams can help minimize subjectivity when identifying hazards and probabilities of occurrence and estimating risk reduction effectiveness.
The quality risk management process owner is responsible for coordinating quality risk management activities across the organization, ensuring the quality risk management process is established, implemented, and maintained, ensuring the adequacy of resources (training and experience), and ensuring subjectivity is controlled to facilitate robust risk-based decision-making based on science and data.
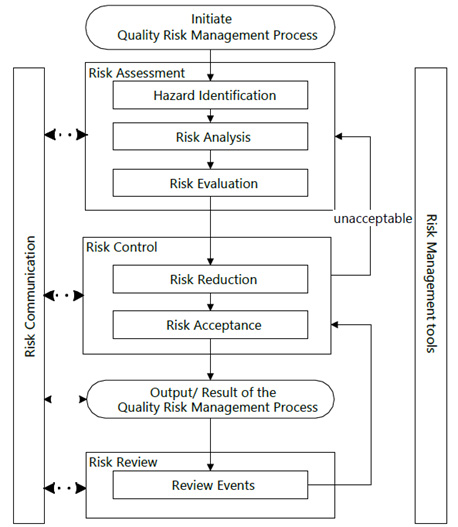
Figure 1: Overview of a typical quality risk management process.
The quality risk management process should be designed to coordinate, facilitate, and improve risk-based decision-making. An effective quality risk management process should use these basic steps:
- define the problem
- form an interdisciplinary team
- identify a leader
- gather the background information
- develop a timeline
- establish deliverables.
Quality risk assessments begin with a well-defined problem description or risk question to identify hazards and analyze and evaluate the risks associated with exposure to the identified hazards. Three useful questions that can be used by the interdisciplinary team to the define the risks include:
- What might go wrong (hazard identification)?
- What is the likelihood it will go wrong (risk analysis)?
- What are the consequences of the severity (risk evaluation)?
Effective risk assessments rely on the quality of the data used to make the risk assumptions. Documenting the rationale for assumptions will aid the current and future team members in the decision-making process. Knowledge gaps related to pharmaceutical sciences, including processing, hinder the interdisciplinary team’s ability to properly identify sources of harm, probability of occurrence, and the ability to detect the issue. Depending on the risk management tools and methods used, risk assessments can provide a quantitative or qualitative estimate of risk. Quantitative risk is expressed as a numerical probability while qualitative risk is described, for example, as high, medium, or low.
The risk control phase involves reducing the risk to an acceptable level while deciding to reduce and/or accept the identified risks. Useful questions that can be used by the interdisciplinary team to control the risks include:
- Is the risk above an acceptable level?
- What can be done to reduce or eliminate risks?
- What is the appropriate balance among benefits, risks, and resources?
- Are new risks introduced as a result of the identified risks being controlled?
Risk reduction focuses on mitigation or avoidance of risk when the risk exceeds a preestablished limit. Risk reduction activities may include reducing the severity of the harm, decreasing the frequency of occurrence, and increasing the ability to detect the hazards associated with the harm. The decision to accept residual risks should be supported with a documented rationale realizing risks may not be fully eliminated.
Sharing of information about risk and risk management between the decision-makers and other interested parties is the basis for an effective risk communication process. Risk communications might include internal company stakeholders, regulators, and industry and industry and the patient and may include information related to the existence, nature, form, probability, severity, acceptability, control, treatment, and detectability of risks.
Risk management should be a continuous ongoing process and contain a mechanism for reviewing and monitoring events, including results of product reviews, inspections, audits, change control, complaints, adverse events, recalls, field actions, new knowledge, and experience.
There are several widely recognized tools and techniques that can be used for risk management, including:
- Basic risk management facilitation methods
- Failure mode effects analysis (FMEA)
- Failure mode, effects and criticality analysis (FMECA)
- Fault tree analysis (FTA)
- Hazard analysis and critical control points (HACCP)
- Hazard operability analysis (HAZOP)
- Preliminary hazard analysis (PHA)
- Risk ranking and filtering
- Statistical tools
The selection of risk management tools depends upon specific facts, circumstances, and the level of experience of the interdisciplinary team. The guidance’s Annex I: Quality Risk Management Methods and Tools provides an overview, references, and potential areas of use for the tools and techniques that might be used by industry and regulators for the risk management process.
Quality risk management can be used as part of an integrated quality management system, including regulatory operations, development, facilities, equipment and utilities, materials management, production, inspection, laboratory control, stability studies, packaging, labeling, supply chain control, storage, and distribution. Q9(R1) Quality Risk Management Annex II: Quality Risk Management as Part of Integrated Quality Management identifies potential uses of quality risk management principles and tools by industry and regulators.
Conclusion
Q9(R1) Quality Risk Management is well written and provides a good basis for establishing an effective risk management program. Additional information about the proposed changes to Q9(R1) Quality Risk Management from the ICH can be found at WG Presentations/Trainings.
Please submit written comments to the Dockets Management Staff, Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852 or electronic comments to https://www.regulations.gov. Please reference docket number FDA-2022-D-0705 with all comments. For questions regarding this draft document, contact Rick Friedman, Rick.Friedman@fda.hhs.gov.
About The Author:
 Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC, an ASQ Fellow, and SRE Fellow. Durivage primarily works with companies in the FDA regulated industries (medical devices, human tissue, animal tissue, and pharmaceuticals) focusing on quality management system implementation, integration, updates, and training. Additionally, he assists companies by providing internal and external audit support as well as FDA 483 and warning letter response and remediation services. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications, including CRE, CQE, CQA, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. You can reach him at mark.durivage@qscompliance.com with any questions or comments.
Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC, an ASQ Fellow, and SRE Fellow. Durivage primarily works with companies in the FDA regulated industries (medical devices, human tissue, animal tissue, and pharmaceuticals) focusing on quality management system implementation, integration, updates, and training. Additionally, he assists companies by providing internal and external audit support as well as FDA 483 and warning letter response and remediation services. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications, including CRE, CQE, CQA, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. You can reach him at mark.durivage@qscompliance.com with any questions or comments.