
How To Break The Pharmaceutical 2-3 Sigma Barrier (Like Amgen)
By Ajaz S. Hussain, Ph.D.
")
A recent U.S. FDA publication entitled The Future of Pharmaceutical Quality and the Path to Get There suggested that the future of pharmaceutical quality is Six Sigma, meaning that no more than 3.4 defects occur per million opportunities (at every manufacturing facility). The way to achieve this goal is to move from the current management standards to performance standards. The need for an additional incentive — the economic driver — was also recognized in the publication. The proposed path forward aims to achieve the long-standing vision of the FDA’s Center for Drug Evaluation and Research (CDER): “a maximally efficient, agile, flexible pharmaceutical manufacturing sector that reliably produces high-quality drugs without extensive regulatory oversight”.1
In seeking a formal mechanism for recognizing the (good) performance of a company and its pharmaceutical quality system (PQS, as indicated in ICH Q10), Six Sigma 3.4 defects per million (dpm) is used as an example. A “pull” vector — a business advantage that would attract others to emulate — is the goal.
On the other hand, it is well known that a segment of the industry is trapped in a “file first, figure it out later” mindset and business model. Some only try to meet standards after they are issued a complete response letter or a warning letter. This is a components of the “push” vector in the current management-based standards, where the FDA pushes (manages) companies to change or be excluded from the U.S. market.
The technical know-how of achieving Six Sigma 3.4 dpm, real-time controls, and continuous manufacturing has existed for many years. Outside the pharma sector, problems that we continue to struggle with have been solved using these tools. So what is the gap between what we know and what we can implement?
Historically the pharmaceutical regulatory system evolved as a reaction to one crisis after another, and over the years each new crisis added a layer of regulatory requirements — which has become the so-called “extensive regulatory oversight,” utilizing management standards to manage a sector that “is unable to manage itself.” The system has become so complex that we often find ourselves in over our heads. As Albert Einstein very simply put it, we will never solve the problems of tomorrow with the same thinking we used to create the problems of today. To elevate our thinking, we must overcome our immunity to change — a concept that discussed in the 2016 CPhI Annual Report.2, 3 This report illustrates how the constructive development theory of human development and the immunity to change concept, developed by Robert Kegen and colleagues at Harvard University, 7 provides a framework for us to objectively fill the gap between what we know and what we can implement in the pharma sector.
Amgen’s Journey To Six Sigma 3.4 dpm
Amgen is a multinational biopharmaceutical company with a biotech and biosimilar product portfolio. From several public presentations by Martin VanTrieste, the company’s former chief quality officer (now retired), we can gain valuable insights into Amgen’s operational excellence journey to Six Sigma, which apparently began in 2005-2006.4-6 (Editor’s note: The author has no affiliation with Amgen.)
Several factors appear to have provided the motivation for Amgen to undertake this voyage: discussion of Six Sigma at the FDA Science Board Meeting on November 21, 2001; the progress achieved in other sectors (e.g., semiconductor and automotive); the September 3, 2003 Wall Street Journal article “New Prescription For Drug Makers: Update the Plants”; and FDA cGMP observations on the inadequacy of out-of-specification (OOS) and deviation investigations. The critical question Amgen leaders posed to themselves was, “Why are investigations important?” The answer: “Stuff happens!” They decided to leave behind the head-in-the-sand mindset and begin their journey by reaffirming the mission to serve patients.
By 2010, Amgen had reduced error rates by 90 percent, scrap by 54 percent, disposition cycle time by 54 percent. Progress was significant, and 65 percent of the company’s manufacturing processes and product parameters were at 6 sigma. At the end of 2014, 82 percent of parameters were performing at 6 sigma, 14 percent were at 3 sigma, and 4 percent had corrective and preventive actions (CAPAs) open to improve performance. Cost savings were also impressive, accumulating to over $300 million. Given the visible progress in reports through 2014, perhaps the company has by now reached the target of 3.4 dpm or less. Constructive Development Theory
What Can We Learn From Amgen’s Journey?
The ICH Q10 model for pharmaceutical quality systems served as a handy guide for Amgen. Management took responsibility, with leaders committing to monitoring manufacturing processes and product attributes using a familiar roadmap to process capability. Leaders focused on poorly performing manufacturing processes and decided that any Ppk (process performance index) lower than 1.0 required a CAPA to drive continuous improvement.
Within the PQS, leaders took a hands-on approach to ensure effective investigations and CAPA. Utilizing PQS enablers, they established risk-based classification, trending nonconformance, and science-based investigations (which must have included knowledge sharing with subject matter experts), and provided regular management review of all aspects of the PQS.
Leaders also focused on of prevention of errors, system-wide, via empowerment (i.e., leaders becoming mentors). Their efforts included, among others, utilizing network-wide metrics to gauge the effectiveness of established controls, standardizing new tools and procedures, and implementing education and training (e.g., familiar operational excellence tools, rigorous scientific investigations, technical writing skills, etc.). A significant emphasis was placed on qualification and then certification of investigators. At a later stage, a focus on mentoring is evident. Prevention of errors and continual improvement were the targets from the beginning. On the factory floor, these efforts were initiated early (perhaps with a short lag after investigations and CAPA) to ensure new errors were not continually being added to the list for investigations.
Throughout this journey, leaders worked to overcome organizational and individual immunity to change. Initially, the leaders confronted their change-prevention system and feelings, and then those of the future leaders (staff). Their emphasis on the knowledge system ensured that new learnings were embodied and day-to-day practices were improved (e.g., developing future leaders at every level, leaders empowering and delegating, etc.). In addition to training, qualification, and certification, operational excellence on the floor was supported by visual knowledge management (information on notice boards), walk the talk (purposeful presence on the floor), work center teams, problem-solving solutions, and learning groups.
In summary, the following was required for Amgen to progress on this journey to 3.4 dpm:
- Analytical characterization of raw materials, manufacturing processes, and products — not just in the development phase but also in the commercial setting (as needed)
- Management involvement in identifying, tracking, and controlling variation via process capability assessment
- Continual monitoring to ensuring robust analytical methods, manufacturing processes, and products (e.g., using industry benchmark for analytical variability and decreasing assay variability)
- Training, qualification, certification, and mentoring support to ensure flawless execution
- A focus on supply chain controls and confidence
This journey is illustrated in Figure 1.
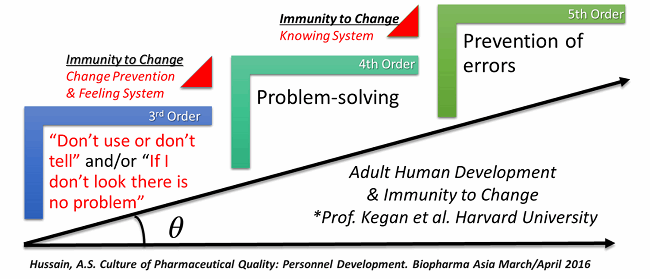
Figure 1: A journey to pharmaceutical six sigma — 3.4 dpm overcoming immunity to change and raising the order of consciousness of the PQS
Applying The Constructive Development Theory To The Pharma Industry
In the constructive development theory, the third order (of consciousness) is the socialized mind.7 Currently, a significant segment of the pharma industry has been socialized, through the power of FDA’s enforcement of management based standards. The most efficient way for products to get to market and remain on the market is to follow FDA guidance and feedback (e.g., complete response letters and warning letters). Unfortunately, such enforcement can diminish motivation within companies to take responsibility for quality.
In Figure 1, the phrase “if I don’t look there is no problem” represents a mindset leaders at Amgen had to overcome to the begin their journey. It took a sustained focus and effort by leadership over a decade to achieve the performance levels they sought. Similarly, the phrase “don’t use or don’t tell” was used by Pfizer at the FDA Science Board Meeting of November 16, 2001 — effectively kicking off the FDA’s process analytical technology (PAT) initiative.
Systems orientation is achieved at the fourth order of development, known as the self-authored mind. Academic self-authorship — as in a doctoral dissertation or a peer-reviewed publication — is a significant development milestone in an academic career. However, in industrial and regulatory settings, self-authorship must also entail an understanding of the part you play in the PQS, how you contribute to its effectiveness, and the responsibility you have for the quality the PQS delivers. An effective systems ordination is essential. For building and maintaining an effective PQS, a critical mass of fourth-order consciousness is necessary for the work environment, with all of its time, costs, and other pressures and stressors. A subject matter expert focused only on his/her narrow technical area of expertise may be fourth order within this field, but only third order within the broader PQS.
The fifth order — the self-transforming mind — requires a prevention mindset that can appreciate multiple systems that are often separated by time and space (e.g., a development system for R&D and a quality assurance system for operations). Based on the available data from developmental psychology and related disciplines, the fifth order is achieved by a tiny percentage of adults — and only when they reach an age when they start to consider retirement. It is unlikely that a critical mass of fifth order consciousness would be present in most companies. We must, therefore, find solutions to align and leverage both the pull and push vectors based on the fourth order, which is consistent with the notion of regulatory science.
Incentivizing Self-Authorship And The Pull Vector
The FDA’s encouragement and support for continuous manufacturing and emerging technologies are abundantly evident. For the FDA to encourage industry to adopt these approaches, the constructive development theory suggests that it should facilitate self-authorship of the related performance standards, which is precisely what the agency is doing.

Figure 2: Do legacy practices constrain the pull vector?
Currently, the FDA lacks a formal mechanism for recognizing corporations such as Amgen that demonstrate the effectiveness of their PQS via performance standards such as Six Sigma 3.4 dpm. However, the incentive for self-authorship of PQS performance standards is emerging rapidly in the biotechnology and biosimilar sector, where the coveted interchangeability designation is still evolving.
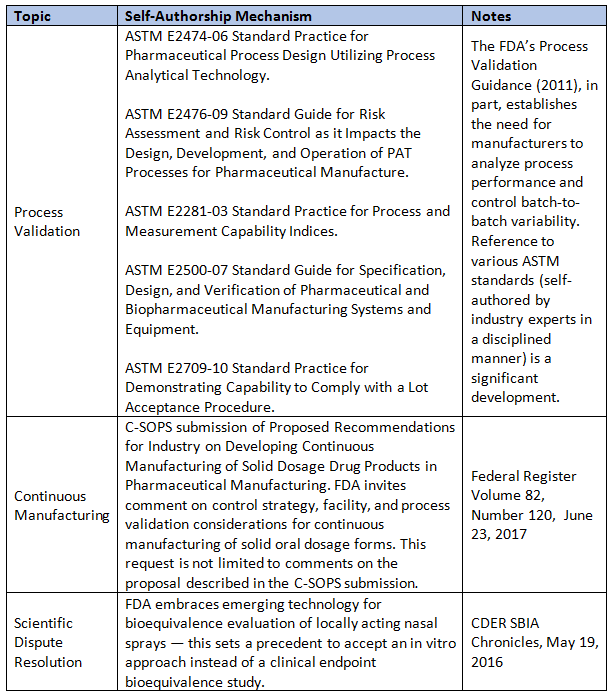
Table 1 below lists several mechanisms that support self-authorship of standards, along with precedence-setting regulatory decisions. A disciplined approach such as the ANSI-accredited standard-setting process — as in ASTM International or the newly credentialed Parenteral Drug Association (PDA) — is necessary to develop objective standards that are in the interest of the broader society. Note: The process of scientific dispute resolution is only listed in Table 1 to recognize that it is made available by the FDA for drug-specific applications, yet it can be precedent setting.
Table 1: Formal Self-Authorship Mechanisms In The U.S. That Industry Can Leverage
Perhaps the FDA’s effort on quality metrics may eventually evolve to provide such a mechanism. However, it is currently struggling to achieve industry consensus.8
The current draft guidance on quality metrics lumps together both biotechnology and synthetic drug products. In doing so, it inadvertently appears to discount the different regulatory legacies (e.g., the role of compendial standards), practices (e.g., team-based cGMP inspections), and recognition of the importance of pharmaceutical equivalence and overall complexity. For example, in the synthetic drug sector, Judge Wolin’s aphorism in the United States v. Barr Laboratories decision— “testing into compliance” — and how compendial standards are utilized (for batch release decisions) can interact to impact the effectiveness of out-of-specification (OOS) investigations adversely.9 It can also convolute the interpretation of the quality metric the FDA is seeking, namely the invalidated OOS rate.
The need for additional self-authoring action by and for the synthetic drug product manufacturing industry is evident, particularly to develop performance standards for what constitutes adequate OOS investigations (e.g., by leveraging FDA referenced standard such ASTM E2709-10 to address the legacy challenges). It could be a stepping stone to a broader set of performance standards such as Six Sigma 3.4 dpm without relying on the continuous manufacturing process.
The Push Vector And The 2-3 Sigma Barrier
In the previous section, we discussed self-authorship as a development stage for the sector and recognized that a disciplined approach is needed to develop objective standards that are in the interest of society. The subjective-to-objective (art-to-science) transformation is at the core of the FDA’s Pharmaceutical Quality for the 21st Century initiative. To improve the objectivity of our decisions on the quality of pharmaceuticals, we must acknowledge that it is confounded within the clinical trials, it is not absolute, and it is multifactorial — it is like an elephant in the dark.10
Assurance of quality, at a very high level, is in part derived from conforming to a set of specifications (attributes, acceptance criteria, and analytical method) for material attributes we know or believe and agree to be critical. The other part from which we derive assurance is the description of the numerous process (i.e., operational routines or standard operating procedures [SOPs]) needed to manufacture the drug product over its commercial lifecycle. Conforming to the SOPs we refer to as “good practices” is based on the principle of “first, do no harm.” It is an important pillar upon which U.S. drug laws and regulations are set; hence, deviations from SOPs can be sufficient to consider a product to be “adulterated.” The principle also speaks to the intention to adhere to good practices. Several tragedies, corrupt intentions, and improving knowledge of science and technology have contributed to the evolution of our collective PQS. Today, it is a highly complex system, global in scope and prone to repeat errors and failures. Tragedies of a magnitude similar to the 1937 sulfanilamide disaster still occur, and the richest nation on Earth struggles to ensure availability and affordability of pharmaceuticals and to provide the assurance patients need consistently. So what is the gap between what we know and what we can implement?
Twelve years ago I suggested that the gap was our inadequate technical education, and particularly training as pertains to measuring and controlling physical attributes of materials and pharmaceutical products.11 The claim is as correct today as it was then. The critical role of adaptive learning — what we learn from experience — was and is severely underestimated in our industry, though it is recognized in, for example, 21 CFR 211.25.12
Adaptive learning differs from technical learning, and we must acknowledge and honor this difference. Adaptive learning occurs in an uncertain and complex environment shaped by a tortuous legacy. This legacy can often serve as a blindfold — thus, the metaphor of six blindfolded men and the elephant is an apt description of the gap we must fill objectively by frequently asking, “How do I know what I know?” Epistemology.
In the current state, FDA’s complete response letters, warning letters, and quality metrics (particularly invalidated OOS rate) are lining up to strengthen the push vector. In doing so, perhaps inadvertently, they may be perpetuating the legacy of the 2-3 sigma barrier, which established the legacy of the generic drug scandal. A sector-level self-authorship action to develop performance standards for what constitutes adequate OOS investigations — e.g., by leveraging FDA-referenced standard such ASTM E2709-10 to address the legacy challenges — can be an important part of the solution. The other and the more critical part is to improve education, training, and experience by certifying epistemology to increase our order of consciousness to self-authorship at every level and in every organization.
Important Implications Of These Insights
Companies with mature PQS such as Amgen (and others who have not yet publicly discussed their performance) should consider self-authoring performance standards to leverage the ongoing transformation. In doing so, these leaders create, simultaneously, a competitive business advantage and the pull vector in their organizations. The evolving biosimilar sector, assurance of interchangeability, and the societal need for a high level of quality assurance are juxtaposed in a way that this self-authoring opportunity may also be a responsibility.
Companies working on maturing their PQS in the absence of FDA warning letters should utilize the insights shared in this article to improve and to accelerate their approach. The journey to establish an effective PQS and to achieve performance such as Six Sigma 3.4 dpm takes years of dedicated commitment and direct management involvement. Companies must prioritize their effort to be proactive in response to the FDA’s effort to strengthen the push vector. Establishment and effective communication of totality of evidence — demonstrating quality by design (QbD) during development, the predictability of scale-up and technology transfer, and stability and capability of analytical methods and processes over their lifecycle — are needed to be proactive. Specific attention must be placed on removing the legacy blindfolds and achieving self-authorship at every level of the organization. It means that operators at the frontline know why they do what they do and can self-author how they do it.
Companies working on maturing their PQS following an FDA warning letters should actively seek to achieve self-authorship at every level of the organization and reduce dependence on external consultants to author policies and SOPs. They must place a heavy emphasis on the effectiveness of root-cause investigations and be informed of the legacy challenges that make such investigations difficult and often incapable of distinguishing between common and special causes. Information about making the distinction between common and special causes is broadly available and in part contained within the ASTM standards cited in the FDA’s Process Validation Guidance (2011). This knowledge should form the foundation of self-authored protocols for investigations and for communicating with FDA's Office of Compliance.
This article set out to answer the question, “What is the gap between what we know and what we can implement?” The answer to this question is found in Einstein’s warning that we will never solve the problems of tomorrow with the same thinking we used to create the problems of today. Utilizing the constructive development theory, a vocabulary and framework was adapted to derive and describe insights useful to guide the development of an effective PQS. The pharmaceutical journey of the 21st century is occurring in a highly polarized sociopolitical environment, thus making it difficult to achieve and maintain a higher order of consciousness without working collectively. I will conclude with the wisdom of Gandhi: We must be the change we wish to see in this world.
This article forms part of the CPhI 2017 Annual Report, which will be released during the CPhI Worldwide event in Frankfurt (October 24-26, 2017).
References:
- Yu, L., and Kopcha, M. "The future of pharmaceutical quality and the path to get there." International Journal of Pharmaceutics (2017).
- Hussain, A. S. “Adherence to the Current Good Manufacturing Practice (CGMP) Regulations in the 21st Century.” CPhI Annual Industry Report 2016: Prospects, Analysis and Trends in Global Pharma. October 2016.
- Hussain, A. S. “The culture of Pharmaceutical Quality: Personnel Development.” Biopharma Asia. March/April 2016.
- Van Trieste, M. “CAPA within the Pharmaceutical Quality System.” ICH Q10 Conference. October 2011.
- Van Trieste, M. “Improving the Quality of Investigations.” PIA Meeting. June 2012.
- Van Trieste, M. “The Journey from Good to Great: Process Monitoring Leads to Improving Product Quality.” 2nd Annual FDA/PQRI Conference. October 2015.
- Kegan, R. In over our heads: The mental demands of modern life. Harvard University Press. 1995. and Kegan, R. and Lahey, L.L. Immunity to change: How to overcome it and unlock potential in yourself and your organization.” Harvard Business Press. 2009.
- Cox, B. “FDA's Kopcha Presses Ahead on Quality Metrics Despite Industry Pushback.” The Pink Sheets. July 2017.
- Kuwahara, SS. “A History of the OOS Problem.” BioPharm international. November 2007.
- Hussain, A. S. “Pharmaceutical quality: Elephant in the Dark or Six Blind Men?” LinkedIn blog posts published on September 8, 2015.
- Hussain, A.S. “The Nation Needs a Comprehensive Pharmaceutical Engineering Education and Research System.” Pharmaceutical Technology. September 2005.
- Hussain, A. S. “Education, Training, Experience & 21 CFR 211.25: Theory, Practice, and Epistemology.” LinkedIn blog post published on March 24, 2016.
About The Author:
 Ajaz S. Hussain, Ph.D., is principal of the consulting firm Insights, Advice & Solutions, LLC. Prior to launching the firm, he was chief scientific officer of Philip Morris International, where he developed tobacco plant-based vaccines and established a rigorous scientific basis for the development of a modified-risk tobacco product. Previously, he was VP and global head of biopharmaceutical development for Sandoz, where he helped the organization establish its leadership position in biosimilars.
Ajaz S. Hussain, Ph.D., is principal of the consulting firm Insights, Advice & Solutions, LLC. Prior to launching the firm, he was chief scientific officer of Philip Morris International, where he developed tobacco plant-based vaccines and established a rigorous scientific basis for the development of a modified-risk tobacco product. Previously, he was VP and global head of biopharmaceutical development for Sandoz, where he helped the organization establish its leadership position in biosimilars.
Before working in industry, Ajaz spent 10 years at the U.S. FDA, most recently as deputy director of the Office of Pharmaceutical Science. His contributions to FDA range from research on specific topics (such as the Biopharmaceutics Classification System Guidance) to global policy development (such as ICH Q8) in the area of quality by design. He launched and led the FDA’s process analytical technology (PAT) initiative. He and his teams were recognized with two FDA Scientific Achievement Awards.
He currently serves as president of the National Institute for Pharmaceutical Technology and Education (NIPTE). He is also a fellow of the American Association of Pharmaceutical Scientists (AAPS) and the Swiss Society of Pharmaceutical Sciences. Ajaz received his bachelor of pharmacy degree from the Bombay College of Pharmacy and an interdisciplinary doctoral degree from the University of Cincinnati.