
5 Important Takeaways From The FDA's Revised Quality Metrics Guidance

By Aravindhan Ramakrishnan, KPMG
On November 23, 2016, the Food and Drug Administration (FDA) published a revised draft guidance for Submission of Quality Metrics Data. The guidance includes significant changes to the earlier quality metrics draft guidance issued by the agency on July 28, 2015.
FDA's Quality Metrics Initiative, announced in April 2013, encourages pharmaceutical firms to embrace continuous improvement and foster a culture of quality by collecting and reporting manufacturing quality data. During the comment period for the 2015 draft guidance, manufacturers acknowledged that the initiative has the potential to transform the way the pharmaceutical industry approaches overall product quality and the reliability of the supply chain. However, manufacturers and participants in ISPE’s Wave 2 Quality Metrics Pilot Program, conducted across 28 companies (83 sites, 60 products), expressed concerns over lack of clarity regarding implementation, timelines, definitions, and evaluation guidelines.
The FDA reviewed these comments from industry and revised the draft guidance in 2016. Following are the most significant changes that can be found in the latest draft guidance:
1. FDA will adopt a phased-in, voluntary rollout starting in January 2018.
Drug makers can submit metrics data by product or site on a quarterly basis. While the program is geared towards finished drug products and API manufacturing, all industry manufacturers may report quality metrics data (e.g., atypical active ingredients and excipient manufacturers).
Establishments engaged in the manufacturing, preparation, propagation, compounding, or processing of a covered drug product, or an API used in the manufacture of a covered drug product, are covered by the guidance. Covered establishments that submit product reports to FDA will be identified as “product reporting establishments” and sites reporting are “site reporting establishments.”
Although the program is voluntary, FDA is encouraging all drug makers to report data. The guidance mentions that “FDA may not be able to accomplish the overall goals of a quality metrics reporting program, as described in the draft guidance, from voluntary reporting alone. If FDA does not receive a large body of data from reporting establishments, the ways in which the Agency can use the information may be limited.”
Key takeaways for manufacturers:
The program is voluntary in 2018, but most manufacturers should still assess their quality systems, update the metrics, and prepare metrics data for submission. As a keystone initiative, quality teams can use this program to revitalize their processes and systems.
Key takeaways for contract manufacturing organizations (CMOs):
Most CMOs may need to prepare metrics by both product and site, and submit the data to the product owners. Contract manufacturers can expect changes to their quality agreements to address these requirements and to comply with the Drug Supply Chain Security Act (DSCSA).
2. Covered establishments can submit product reports or site reports.
FDA intends to accept either product- or site-level reporting data from covered establishments. However, the agency encourages product reporting because it demonstrates a further level of oversight and control over the manufacturing of drug products across the supply chain.
Key takeaways for manufacturers and CMOs:
Product owners need to decide if they are going to be product reporting or site reporting establishments. Gathering the metrics by product across the supply chain will be more complex than the site reports. For example, a product owner might be using a CMO who in turn uses a subcontractor, and the API might be supplied through an entirely different entity. Metrics gathering and reporting through an automated system would not be easy between site and companies. Also, the data might reside in multiple manufacturing execution systems (MES), laboratory information systems (LIMS), complaints management systems, and enterprise resource planning (ERP) systems, at the least.
By taking a three-phased approach toward redesigning their quality metrics dashboard and management review processes, manufacturers can drive consistency across the enterprise and partners:
- Determine dashboard KPIs and standardize definitions
- Conduct gap assessment of quality source systems
- Implement system to aggregate data for the dashboard
In addition, the following guidelines can help with the overall program:
- The quality system must include policies and procedures that define the level of management visibility to be enabled by the dashboard and that validate information is flowing to the dashboard as intended.
- All KPIs in the dashboard should include clear definitions with data standards and training for individuals manually entering source data, and should align with FDA guidance.
- Splitting the dashboard into operational quality (e.g., out-of-specs, change controls) vs. post-marketing quality (e.g., field reports) provides a more complete view of performance and risk.
- Include KPI tiers with index KPI(s) that cascade to lower-lever tier metrics to provide differing levels of analysis.
- Accountability and a set cadence should be determined for dashboard refreshes, dissemination, and review.
- KPI targets should be regularly reviewed and trending assessments should be conducted to identify underperformance relative to targets in order to drive action plans.
3. Metric areas and metric definitions have been modified.
Based on the industry comments to the previous draft, the FDA has reduced the scope of the program from four primary metrics and three optional metrics (in the 2015 guidance) to three primary metrics. Those three are:
- Lot acceptance rate (LAR)
- Product quality complaint rate (PQCR)
- Invalidated out-of-specification (OOS) rate (IOOSR)
Furthermore, the FDA has revised the metric definitions as detailed below
Table 1: Revised Quality Metric Guidance Definitions
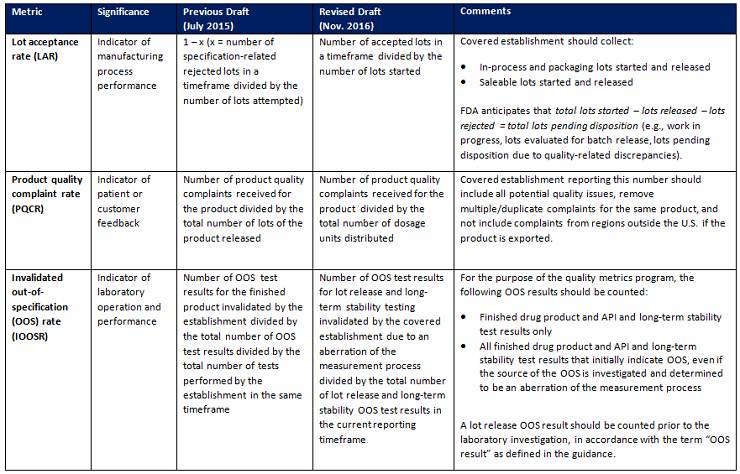
Key takeaways for manufacturers and CMOs:
Covered entities will need to review their metrics management systems across the manufacturing supply chain, identify gaps in data collection, standardize KPIs, review CMOs/subcontractor metrics data, collate and calculate metrics, review metrics with management, and ensure alignment with metrics program guidelines. Standardization is the key to the success of the program.
4. Products imported/manufactured outside the U.S. get special considerations.
Pharmaceutical manufacturing operations and supply chains are very complex, with each drug product often involving numerous sites and multiple contract manufacturing (CMOs) partners. This has significant implications on how quality metrics are collated, calculated, and reported.
Further, some reporting establishments ultimately will be responsible for submitting data for a large number of products. Thus, reporting establishments should consider the following while calculating the three metrics indicated in the new draft guidance:
- LAR and IOOSR — FDA recognizes that it may not be possible for some covered establishments to identify started lots, rejected lots, and OOS results that are manufactured by CMOs outside of the U.S. and then imported. The report can include data from lots not imported along with data from lots imported, provided they follow the same process and controls.
- PQCR — Product quality complaint data should be related to drugs that are imported, intended for import, or manufactured in the U.S.
- PQCR for OTC products — Calculations should exclude preferential complaints.
5. FDA will establish a portal for the submission of metrics data and recognize the reporters.
FDA intends to open an electronic portal in January 2018 for voluntary submissions of quality metrics data on a quarterly basis. A revised version of the Quality Metrics Technical Conformance Guide will be released soon, followed by the release of guidance about the submission portal by December 2017.
Reporting establishments may include up to 300 words of comment with their submissions, mainly to indicate reasons for any significant variations in the data or anything else of importance to note. The submission of comments is optional. In the future, FDA may consider establishing a set of codes to standardize the comments.
For any other clarifications about submitting metrics, reporting establishments may email their questions to OPQ-OS-QualityMetrics@fda.hhs.gov.
To recognize proactive engagement, transparency, and commitment to quality, the agency will identify participating establishments on its website in following categories:
- For product reporting establishments
- Product reporter top tier: Supplied complete data supporting all metrics for each covered establishment in the manufacturing supply chain for all covered drug products or APIs for the full year
- Product reporter mid-tier: Identified all covered establishments in the manufacturing supply chain for all covered products, and provided complete quality metric data from at least one establishment for each covered drug products or API for the full year
- Product supply chain reporter: Identified all covered establishments in the manufacturing supply chain for all covered drug products or APIs
- For site reporting establishments
- Site reporter top tier: Included complete data supporting all metrics for all covered drug products or APIs for the full year
- Site reporter mid-tier: Included complete data supporting all metrics for at least one covered drug product or API manufactured at an establishment for the full year
Key takeaways for manufacturers and CMOs:
Currently, FDA has to sift through fragmented, disparate, and incompatible data sources to understand quality. Data from NDA field alert reports (FARs), recall alerts (RAs), biological product deviation reports (BPDRs), Medwatch 3500A forms, CMC supplements, annual reports, complaints, establish inspection reports (EIRs), and 483s is helpful but not always a clear indicator continuous improvement. With the new guidance, manufacturers can report quality and show progress over time in an easier format that is comparable across the industry.
Pharmaceutical quality — especially among peers — has not always been readily transparent. Compared to other industries such as automotive and aerospace, there is no ready mechanism available for sharing quality data. This would serve as an opportunity for companies to have a hard look at their reporting mechanisms and quality systems maturity.
Conclusion
The pharmaceutical industry is under intense pressure and scrutiny from regulators, customers, stakeholders, and other groups. To produce quality medicines at reasonable cost — and manage supply and demand complexities — manufacturers will need to run their operations vastly different than they do today.
In a recent report, FDA’s Office of Pharmaceutical Quality stated grimly that it has “only limited information about the current state of pharmaceutical quality.” The success of the quality metrics reporting program is the bedrock to FDAs grand vision for building quality into drug products. To help achieve this vision, the agency needs the industry to shift from a culture of rules and regulations to a culture of quality, where quality is embedded in every process.
With its new quality metrics draft guidance, FDA is nudging manufacturers to change. It will be interesting to see how the manufacturers respond. This is an opportunity for pharmaceutical manufacturers to see these changes not simply as a new set of rules, but as an opportunity to demonstrate their leadership in how they manage the quality of their products.
About The Author:
 Aravindhan (Arvi) Ramakrishnan is a manager within KPMG’s Life Sciences Advisory Practice, with over 12 years of experience. He focuses primarily on helping clients comply with various global compliance requirements, quality management systems, and large transformation programs. Prior to joining KPMG, he was in a global consulting role working with clients across Europe and the U.S. in strategy, operations, supply chain, quality management, and R&D. You can reach him at aravindhanramakrishn@kpmg.com.
Aravindhan (Arvi) Ramakrishnan is a manager within KPMG’s Life Sciences Advisory Practice, with over 12 years of experience. He focuses primarily on helping clients comply with various global compliance requirements, quality management systems, and large transformation programs. Prior to joining KPMG, he was in a global consulting role working with clients across Europe and the U.S. in strategy, operations, supply chain, quality management, and R&D. You can reach him at aravindhanramakrishn@kpmg.com.