
Introduction To Science- And Risk-Based Cleaning Validation Using ASTM E3106 & E3219
By Andrew Walsh, Thomas Altmann, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G. Dolan Ph.D., Andreas Flueckiger, M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle, Ph.D., Ovais Mohammad, Mariann Neverovitch, and Osamu Shirokizawa
Part of the Cleaning Validation For The 21st Century series
This article discusses some of the history that began the movement from compliance-based approaches to cleaning validation to the science- and risk-based approaches introduced in the American Society for Testing and Materials (ASTM) E3106 Standard Guide for Science Based and Risk Based Cleaning Process Development and Validation and the ASTM E3219 Standard Guide for Derivation of Health Based Exposure Limits (HBELs).
Historical Developments Affecting Cleaning Validation
Historically, the pharmaceutical industry has mainly approached cleaning validation as a compliance exercise. As such, cleaning validation activities were generally established based on regulatory expectations from observations during inspections and not on science-based master plans or risk assessments. In the early 1990s, the FDA, as well as other regulatory agencies, began to view cleaning as a process that required validation.1 So, cleaning validation became closely associated with process validation.
Cleaning validation was based on the traditional process validation approach, which uses preapproved protocols, with predetermined acceptance criteria and the standard three runs. This approach was adopted without considering if three cleaning validation runs were needed or sufficient or if predetermined acceptance criteria were appropriate for the validation of cleaning. At the same time, the industry was struggling over how to set these required predetermined acceptance criteria.
As part of the FDA’s GMPs for the 21st century project, starting around 2001, many new initiatives came from regulatory agencies and from the pharmaceutical industry itself. Three significant events that occurred prior to this time also shaped the direction of the pharmaceutical industry and of cleaning validation – the Barr Labs decision in 1993, the FDA's 1993 Guidance on Cleaning Validation, and the 1996 proposed revisions to the GMPs.
Table 1: Historical Developments Affecting Cleaning Validation
|
Organization |
Year |
Event / Initiative |
|
FDA |
1993 |
United States vs. Barr Laboratories2 |
|
FDA |
1993 |
FDA Guidance for Inspection of Cleaning Processes1 |
|
FDA |
1996 |
Proposed Revisions to the GMPs3 |
|
Industry |
~2000 |
Operational Excellence (Lean Manufacturing / Six Sigma) |
|
FDA |
2004 |
Pharmaceutical CGMPs for the 21st Century Industry – A Risk-Based Approach4 |
|
FDA |
2004 |
FDA Guidance for Industry: PAT5 |
|
EMEA |
2005 |
EMEA Concept paper on dedicated facilities for certain products 6 |
|
ICH |
2005 |
Quality Risk Management – Q97 |
|
ICH |
2008 |
Pharmaceutical Development – Q88 |
|
ISPE/FDA |
2010 |
Risk-MaPP9 |
|
FDA |
2011 |
FDA Guidance for Industry: Process Validation - General Principles and Practices10 |
|
EMA |
2014 |
EMA Guidance on Using HBELs in Shared Facilities11 |
|
ASTM |
2018 |
E3106 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation12 |
|
ASTM |
2020 |
E3219 Standard Guide for Derivation of Health Based Exposure Limits (HBELs) 13 |
Initiatives coming from the pharmaceutical industry included lean manufacturing, Six Sigma, and operational excellence (OpEx), which grew out of the pressures to reduce costs or speed up production and to better supply the market with medicines. These market pressures pushed the industry toward more effective and efficient processes that would focus efforts and resources where they provided the most value. The initiatives from regulators have been focused on improvements to product quality in process development and manufacturing and the use of risk assessments. While the initiatives from the pharmaceutical industry and regulators may appear to be distinct from each other, they can be combined very naturally.
All of these initiatives have been drawing the industry toward science-based, risk-based, statistics-based, and cost-effective approaches to ensuring patient safety and product quality during pharmaceutical development and manufacturing. As cleaning is one of the critical processes in manufacturing, its performance and validation could also benefit from all these initiatives.
The following is a brief discussion14 of the main events that have influenced the current direction of cleaning validation.
United States Vs. Barr Laboratories
During the early 1990s, multiple inspections of Barr Laboratories, Inc. by the FDA resulted in repeated observations. In frustration, Barr Laboratories brought a lawsuit against the FDA. The FDA responded by petitioning the court for an injunction against Barr Laboratories. The outcome of this legal battle was the now famous Barr Labs decision, in which Judge Alfred M. Wolin found merit in all the FDA's claims against Barr Laboratories and agreed that process validation and cleaning validation are required by the GMPs. By extension, the Barr Labs decision applied to the entire industry. At the same time, Judge Wolin criticized the GMPs for being vague and sympathized with Barr Laboratories’ complaints about the FDA's apparently capricious and unpredictable enforcement.15
Eli Lilly Article On Cleaning Limits
At the same time as the Barr Laboratories lawsuit, most pharmaceutical companies were struggling to provide limits for cleaning validation that were acceptable to the FDA. In 1992, the Pharmaceutical Manufacturing Association conducted a survey of its members and found 44 different approaches being used.16 Many limits were being set as a fraction of a pharmaceutical dose or as some concentration level in a batch (e.g., ppm). Then in 1993, two scientists at Eli Lilly and Co. published an article17 describing an approach that included:
- No more than 0.001 dose of any product will appear in the maximum daily dose of another product
- No more than 10 ppm of any product will appear in another product
- No quantity of residue will be visible on the equipment after cleaning procedures are performed
The Eli Lilly approach combined the dose and concentration criteria and was adopted by many companies mostly because the FDA mentioned it in its cleaning validation guidance,1 which was written in response to Judge Wolin's criticisms. Other regulatory agencies quickly adopted these criteria in their own guidance. Eventually, though, many companies found these criteria challenging to comply with and began turning to less stringent approaches, such as 0.01 of a dose or 100 ppm.
FDA 1996 Proposed Revisions To The GMPs
In response to Judge Wolin's criticisms and ongoing industry complaints, the FDA proposed changes to the GMPs in 1996.3 These changes were very clear and specified validation activities such as blend uniformity testing, mixing validation, and analytical method validation. In addition, the FDA specified that it would now expect that, in addition to penicillin, certain "classes" of compounds – such as cytotoxic agents or other antibiotics – would need to be manufactured in dedicated facilities. In the preamble to the proposed changes, the FDA stated:
"The agency has refrained from establishing a list of drugs or drug products that present such an unacceptable risk, because such a list would quickly become obsolete.... FDA expects manufacturers to identify any drugs that they produce that present the risk of cross-contamination and to implement measures necessary to eliminate that risk. FDA recognizes that, depending on the drug product, a variety of measures may be acceptable to eliminate cross-contamination; there may, however, be situations in which nothing short of dedicated facilities or equipment will be sufficient".
While the industry began complying with these proposed validation requirements, the difficulties in complying with the requirement for potential dedicated facilities began to increase. As mentioned above, many companies were already having trouble implementing the Eli Lilly criteria (e.g., very low limits for low-risk products).
ICH Q9 Guidance
The issuance of ICH Q9 provided basic principles of quality risk management (QRM) and examples of tools for QRM that could be applied to pharmaceutical processes. Although successfully used in many other industries prior to ICH Q9, risk analysis was not common in pharmaceutical manufacturing. ICH Q9 contains two primary principles of quality risk management:
- The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient; and
- The level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk.
ICH Q9 also offered a framework for implementing a quality risk management process (Figure 1).
|
|
|
Figure 1: Overview of a typical quality risk management process in ICH Q9 |
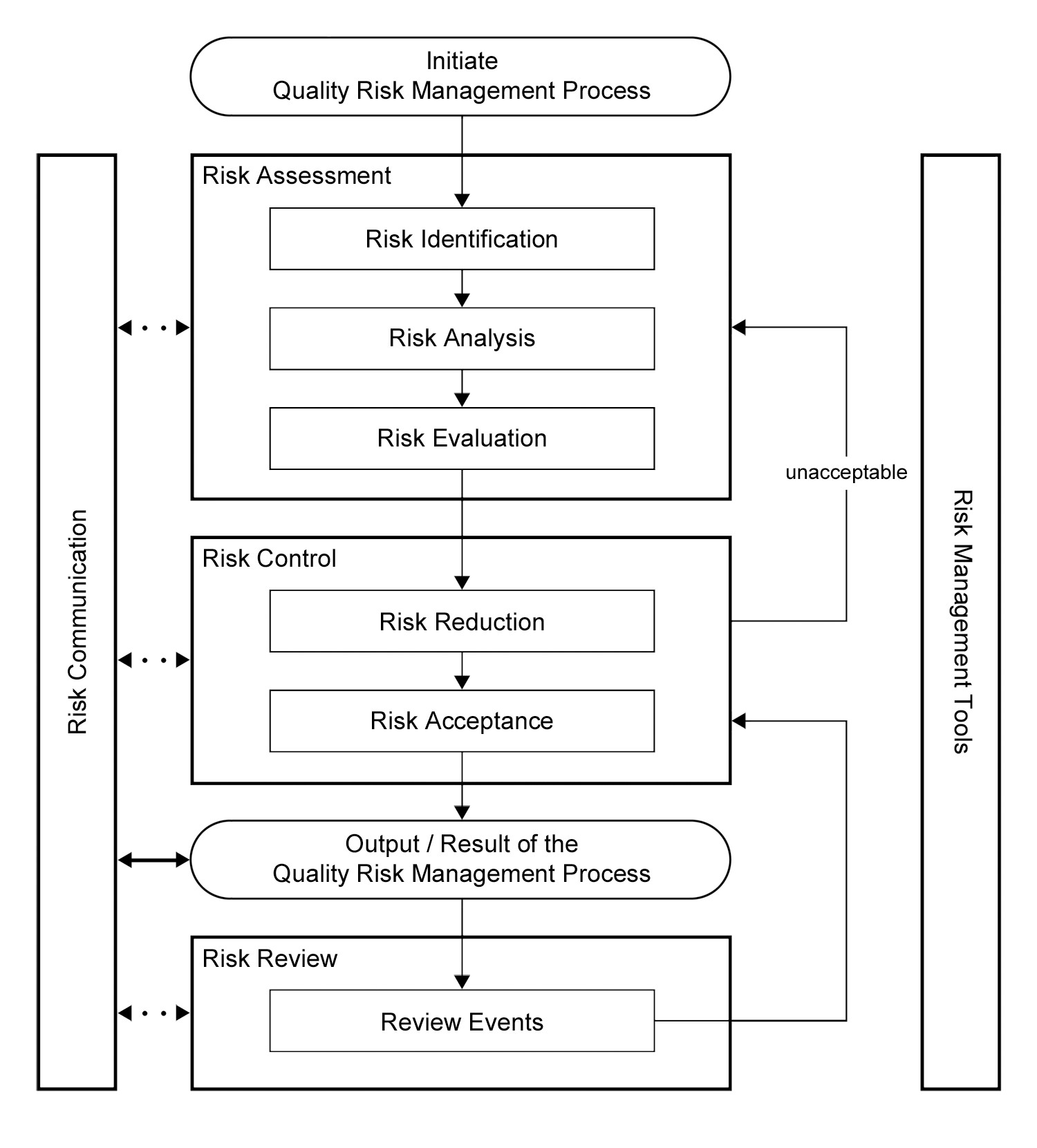
Comparing the ICH Q9 guidance to the FDA's 1996 revision to the GMPs, we can see an opportunity to utilize the two primary principles to address whether the 1996 proposed requirement for dedication of equipment and/or facilities for a product would be necessary.
Risk-MaPP
As the concerns of complying with the dedication requirements began to increase, in 2004 a team of pharmaceutical toxicologists, industrial hygienists, quality assurance professionals, a cleaning validation SME, and a representative from the FDA was formed to create a guide for publication by the International Society for Pharmaceutical Engineering (ISPE). Several of the authors of this article participated in this effort. The intention of this guide was to help companies perform a risk assessment of cross contamination, on a case-by-case basis, to determine the controls needed to ensure patient safety and product quality. This case-by-case approach was intended to avoid the possibility of dedicated facilities based simply on classes of products (e.g., all cytotoxics). This guideline was published in 2010 and is known as the Risk-Based Manufacturing of Pharmaceutical Products, or Risk-MaPP. Essential to the Risk-MaPP approach is an appropriate starting point for the evaluation of cleaning processes.
Over time, the Eli Lilly approach for setting cleaning validation limits was found to lack relevant toxicological information, which resulted in setting lower limits for some low-risk compounds than for high-risk compounds.18 These discrepancies led to a confusing picture of the actual risk of cross contamination and could possibly even result in low-risk compounds (e.g., low-dose aspirin) requiring dedication. Risk-MaPP introduced the concept of an acceptable daily exposure (ADE) as the starting point for a risk assessment that could evaluate the risks of cross contamination for a product and decide whether dedication is necessary. Risk-MaPP defined the ADE as:
"a dose that is unlikely to cause an adverse effect if an individual is exposed, by any route, at or below this dose every day for a lifetime."
The ADE approach is an extremely stringent criteria and initially caused concern, considering that many companies had already found it difficult to achieve the Eli Lilly approach, and this new ADE approach might be even harder to achieve. However, a comparative analysis of the ADE to the 0.001 dose criterion for 304 drug compounds found that the use of the 0.001 dose criterion resulted in the limits for 85 percent of these compounds being set too low, and by a significant degree. On the other hand, the Eli Lilly approach had not been restrictive enough for 15 percent of these compounds.19 So, the initial concerns about the ADE were unfounded and the ADE approach clearly provides a relief for many companies struggling with the Eli Lilly approach. Above all, the approach based on ADEs allows for a risk assessment that shows the true objective safety margins achieved in a product changeover situation.
The ADE was an obvious replacement for the Eli Lilly approach, as it is a health-based value scientifically derived from all the available toxicological and clinical data for the compound and can be used for setting appropriate limits for cleaning and also for worker exposure. Figure 2 shows an overview of the Risk-MaPP process.
|
|
|
Figure 2: Overview of the Risk-MaPP process |
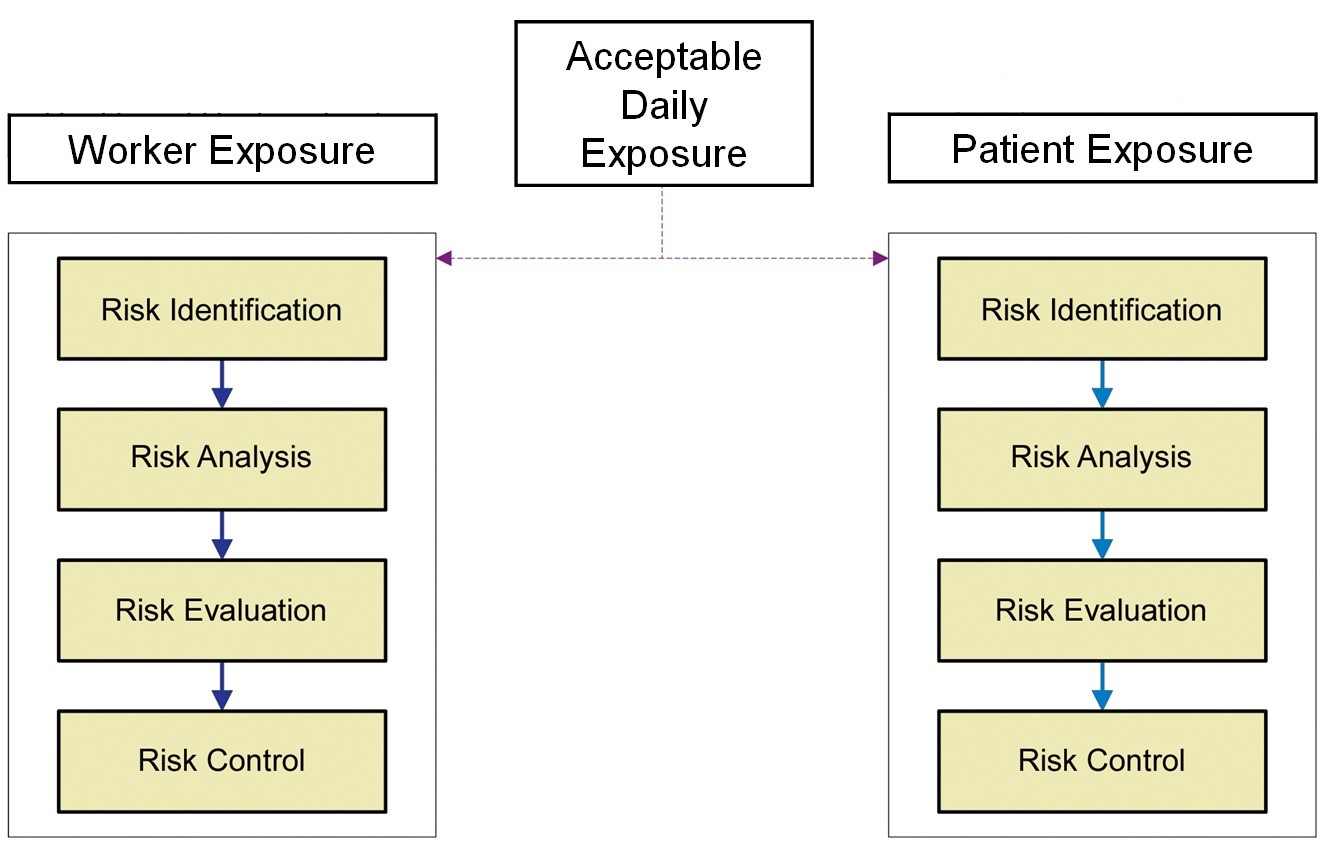
EMEA Concept Paper On Dedicated Facilities For Certain Products6
In its concept paper in 2005, the European Medicines Agency (EMEA) announced that it would make clear what other medicines would require dedicated facilities, in addition to potent sensitizers. The concept paper was mostly concerned with so-called cytotoxics. The industry was very opposed to this proposal, as it conflicted with the quality risk management principles of ICH Q9 which had been issued in the same year and could also lead to many classes of compounds having to be manufactured in dedicated facilities.
FDA 2011 Process Validation: General Principles And Practices
In 2008, the FDA released a draft of its updated process validation guidance10 to align with the product life cycle concept and with existing FDA guidance on ICH Q8-Q10. This new guidance also described concepts that are directly applicable to cleaning and cleaning validation. While this new guidance does not specifically address cleaning, the elements of the process validation guideline can be easily framed as a science-, risk-, and statistics-based approach to cleaning. The FDA has also stated publicly that the new guidance applies to cleaning validation.20
EMA Guidance On Using Health-Based Exposure Limits In Shared Facilities
In November of 2014, the EMA issued a guideline requiring that companies review and evaluate pharmacological and toxicological data of individual active substances to determine their health based exposure limits (HBELs) using permitted daily exposures (PDEs) for use as a risk identification tool and to justify carryover limits used in cleaning validation.10 The EMA uses the term PDE to describe an HBEL and states explicitly that its PDEs are considered equivalent to the ADEs. In this guidance, the EMA required all companies to have HBELs in place by December of 2015.
The requirement to use HBELs was also incorporated into Annex 15 in October 2015 and the previous references to using 0.001 dose and 10 ppm for cleaning validation were removed.21 The Pharmaceutical Inspection Co-operation Scheme (PIC/S), which now includes 54 health authorities around the world, including the FDA, integrated EMA’s Annex 15 into its own document series.22 In 2018, PIC/S also adopted literally the GMP guidelines that the EMA had modified in 2014 where the concept of science-based limit values was introduced. So, in effect, continued use of the 0.001 dose and 10 ppm for cleaning validation acceptance limits is no longer recognized as appropriate or acceptable throughout the EU, the U.S., and most of the world.
ASTM Standards And Their Significance
ASTM International is a global organization providing fully transparent development of voluntary consensus standards that has over 30,000 members from more than 140 countries. ASTM members participate on teams to develop standard guides, practices, methods, and specifications using their global expertise, science, and engineering to improve performance in manufacturing and materials, products and processes, systems, and services. ASTM members include businesses, governments, and interested individuals who collaborate openly and transparently on technical committees to develop standards. All standards are voted on by the members of the governing committee and all negative votes must be resolved satisfactorily with the voter until consensus is reached. ASTM currently has over 12,500 standards being used worldwide. ASTM formed the Committee E55 on the Manufacture of Pharmaceutical Products in 2003, and several standards relating to cleaning validation have since been published or are being developed. As one of the few consensus standard developing organizations, ASTM satisfies the requirements of the U.S. National Technology Transfer and Advancement Act of 1995, so the FDA may recognize and adopt ASTM standards.
ASTM E310612
This new standard applies the life cycle approach to cleaning process validation, which includes the development, qualification, and verification of cleaning processes. It is applicable to pharmaceuticals (including active pharmaceutical ingredients [APIs], dosage forms, over-the-counter drugs, veterinary products, biologics, and clinical supplies) and is also applicable to other health, cosmetic, and consumer products. This standard utilizes the HBEL (ADE) for evaluating the risk to patients from the cleaning of manufacturing equipment and medical devices and will be aligned with the E3219 standard.
|
|
|
Figure 3: ASTM E3106 cleaning risk management process |
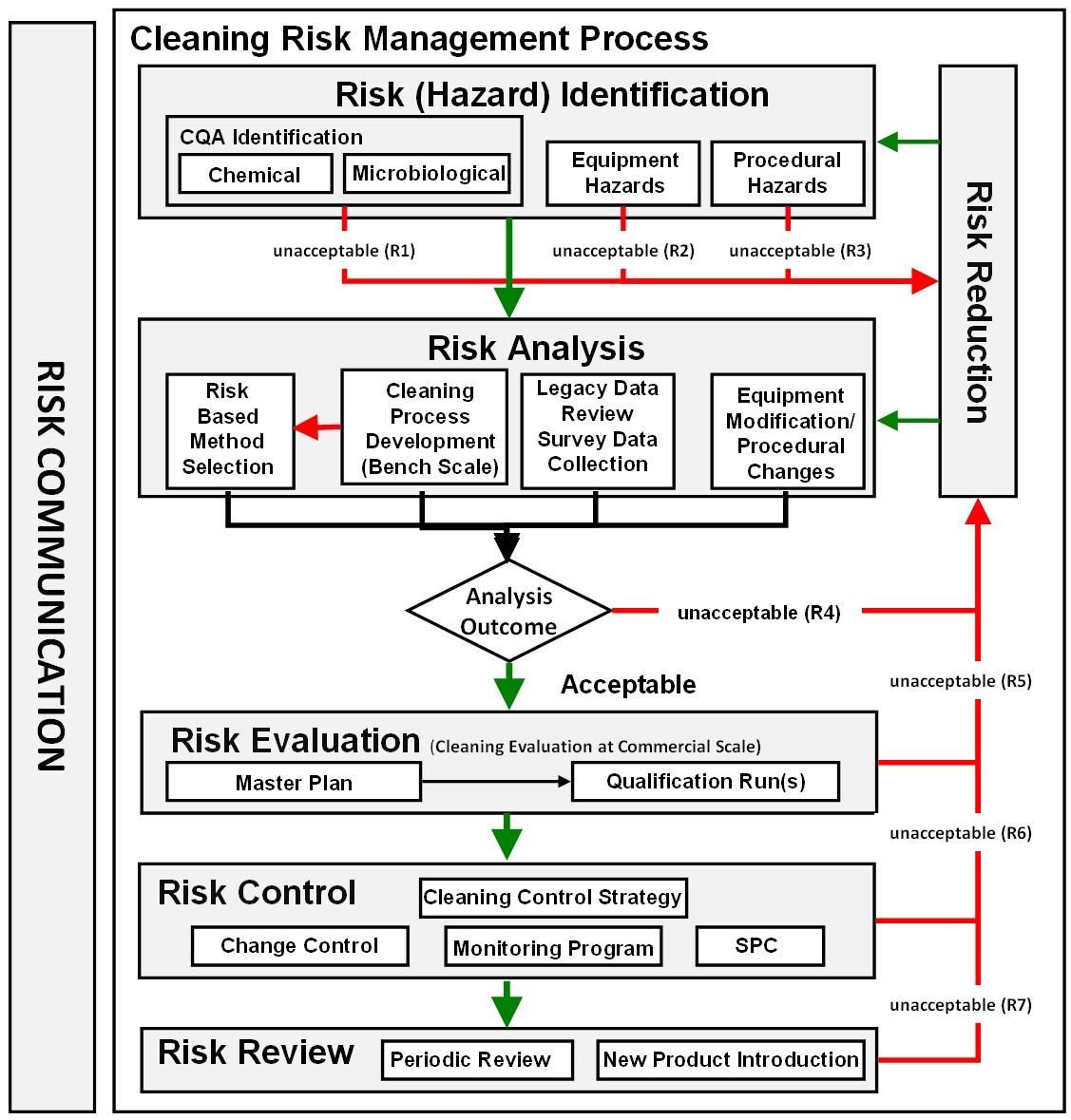
The new ASTM E3106 focuses much more attention on applying science and risk at the risk identification and risk analysis stages, including cleaning process development, analytical method selection, risk analysis of legacy data, risk analysis of SOPs, etc. (Figure 3). Appropriate efforts at these early stages can provide reductions in the level of effort, formality, and documentation of the overall validation process, allowing the selection of risk-based analytical methods (e.g., visual inspection) and simplifying the introduction of new products.
ASTM E321913
The purpose of E3219 is to standardize the derivation of the HBEL. Both Risk-MaPP and the EMA's Guideline on Health Based Exposure Limits contain limited guidance and their calculations are different in some respects. E3219 describes the scientific procedures for evaluating and interpreting the toxicological and clinical data for an API and how to utilize the data to derive an HBEL that can be used for evaluation of cross contamination during the manufacture of different products in the same manufacturing facilities. E3219 should be used for calculating and documenting an HBEL for APIs (including biologics), intermediates, cleaning agents, excipients, or other chemicals that have been identified as hazards for cleaning. E3219 references, and is meant to be used in conjunction with, the ASTM E3106 Standard Guide.
|
|
|
Figure 4: ASTM E3219 example of process for final selection of an HBEL (Reprinted from ASTM E3219-20 Standard Guide the Derivation of Health Based Exposure Limits (HBELs), copyright ASTM International, 100 Barr Harbor Drive, West Conshohocken, PA 19428, USA, www.astm.org.) |
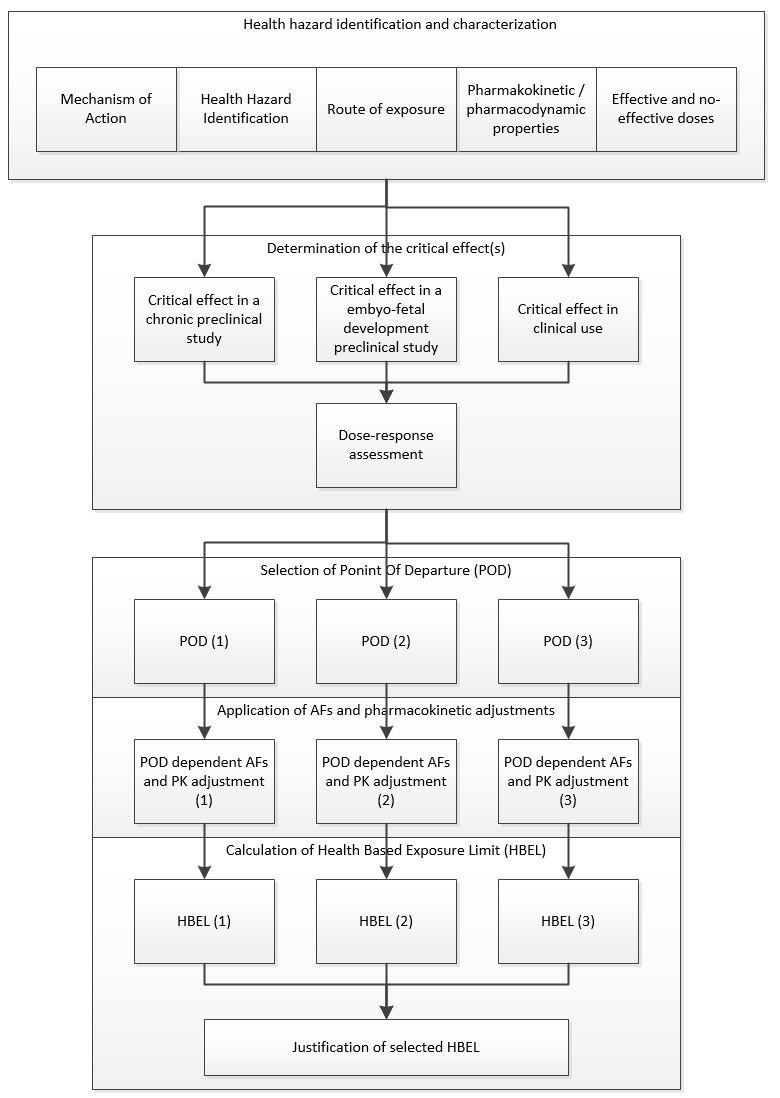
E3219 also defines the qualifications that are required of persons deriving the HBEL (qualified expert) and the minimum requirements for documentation of the derivation. The standard is also intended to be used by regulatory inspectors (e.g., FDA, EMA, etc.) for evaluating the HBELs they find during inspections.
ASTM Standards In Development
There are currently several additional ASTM standards in various stages of development on important topics in cleaning validation. Two of these are ASTM work item 64938 and ASTM work item 67425.
ASTM work item 64938, Standard Practice for the Calculation of Cleaning Validation Limits, is intended to provide guidance on how to use ASTM E3106, Standard Guide Science-Based and Risk-Based Cleaning Process Development and Validation, in combination with ASTM E3219, Standard Guide for Derivation of Health Based Exposure Limits (HBELs), to calculate HBELs for use in the risk assessment of cleaning processes. The calculation of limits for use in cleaning validation has been a source of great confusion for many years and there has been limited guidance on these calculations. This standard will provide detailed guidance on how cleaning limits for pharmaceutical dosage forms, APIs, cosmetics and medical devices should be calculated from HBELs. The calculation of statistical process control limits for cleaning validation will also be discussed.
ASTM work item 67425, Standard Practice Guide for the Qualification of Visual Inspection of Pharmaceutical Manufacturing Equipment and Medical Devices for Residues, addresses visual inspection, which has been used for many years to release manufacturing equipment and medical devices for use after cleaning in the pharmaceutical, biologics, and medical device industries. However, visual inspection has never been demonstrated to be effective, reliable, or safe to use. Recently, the EMA issued a Q&A to its Guideline on Health Based Exposure Limits where it described what criteria must be met for visual inspection to be acceptable to the EMA for release of manufacturing equipment.23 Consequently, a standard is needed to guide these industries in how to demonstrate to the inspectors that they are capable and qualified to accurately assess the absence or presence of residues on manufacturing equipment or medical devices. The scope of this standard will include pharmaceuticals (including APIs, dosage forms, over-the-counter drugs, veterinary products, biological drugs, and clinical trial material) as well as medical devices. It will also be applicable to other health, cosmetics, and consumer products.
Summary
The new ASTM standards discussed above can be utilized to create science- and risk-based approaches to cleaning and cleaning validation. These standards offer guidance for making data-driven changes in the approaches to cleaning validation that can reduce the complexity, lower the costs, and shorten the processes while providing a higher probability that cleaning of pharmaceutical manufacturing equipment is effective. By implementing a truly science-based approach, such as with the use of the HBELs for risk analysis, with appropriate risk assessments, and with cleaning process development in place, a streamlined cleaning program may be readily developed that ensures patient safety and product quality while lightening the regulatory burden on industry. The next articles in this series will provide more detailed discussion of E3106 and E3219.
Peer Review:
The authors wish to thank our peer reviewers: Bharat Agrawal, James Bergum, Ph.D., Sarra Boujelben, Gabriela Cruz, Ph.D., Mallory DeGennaro, Parth Desai, Laurence O'Leary, Tri Chanh Nguyen, Miquel Romero Obon, Siegfried Schmitt, Ph.D., and Joel Young for reviewing this article and for providing insightful comments and helpful suggestions.
References:
- FDA Guide to Inspections Validation of Cleaning Processes, Section IV. Evaluation of Cleaning Validation, July 1993, U.S. Food and Drug Administration (FDA), www.fda.gov.
- United States vs. Barr Laboratories, Inc. Civil Action No. 92-1744, U.S. District Court for the District of New Jersey: 812 F. Supp. 458. 1993 US Dist. Lexis 1932; 4 February 1993, as amended 30 March 1993.
- Current Good Manufacturing Practice: Proposed Amendment of Certain Requirements for Finished Pharmaceuticals. Federal Register / Vol. 61 No. 87 / Friday, May 3, 1996 / Proposed Rules
- Pharmaceutical CGMPs for the 21st Century Industry – A Risk-Based Approach: Final Report, September 2004, U.S. Food and Drug Administration (FDA), www.fda.gov.
- FDA Guidance for Industry: PAT – A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance, September 2004, U.S. Food and Drug Administration (FDA), www.fda.gov.
- EMA Concept paper dealing with the need for updated GMP guidance concerning dedicated manufacturing facilities in the manufacture of certain medicinal products. Doc. Ref. EMEA/152688/04. Feb 2, 2005
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline, Quality Risk Management – Q9, Step 4, 9 November 2005, www.ich.org.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline, Pharmaceutical Development – Q8(R2), August 2009, www.ich.org.
- ISPE Baseline® Guide: Risk-Based Manufacture of Pharmaceutical Products (Risk-MaPP), International Society for Pharmaceutical Engineering (ISPE), First Edition, September 2010.
- FDA Guidance for Industry: Process Validation - General Principles and Practices January 2011, U.S. Food and Drug Administration (FDA), www.fda.gov.
- EMA Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities, EMA/CHMP/CVMP/ SWP/169430/2012, 20 November 2014.
- American Society for Testing and Materials E3106 "Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation" www.astm.org.
- American Society for Testing and Materials E3219 "Standard Guide for Derivation of Health Based Exposure Limits (HBELs)" www.astm.org.
- For a detailed discussion see: Sargent, E.V., et al., The regulatory framework for preventing cross-contamination of pharmaceutical products: History and considerations for the future, Regulatory Toxicology and Pharmacology (2016), http://dx.doi.org/10.1016/j.yrtph.2016.05.029
- Jimenez, F.J., “Enforcement of the Current Good Manufacturing Practices for Solid Oral Dosage Forms After United States v. Barr Laboratories” 52 Food Drug L.J. 67, 1997
- Pharmaceutical Manufacturing Association survey on 'Cleaning Validation" 1992.
- Fourman, G., and Mullin, M., “Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations,” Pharmaceutical Technology, April 1993, www.pharmtech.com
- Walsh, A., “Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part I,” Pharmaceutical Engineering, July/August 2011, Vol. 31, No. 4, pp. 74- 83.
- Walsh, Andrew, Michel Crevoisier, Ester Lovsin Barle, Andreas Flueckiger, David G. Dolan, Mohammad Ovais (2016) "Cleaning Limits—Why the 10-ppm and 0.001-Dose Criteria Should be Abandoned, Part II," Pharmaceutical Technology 40 (8)
- International Pharmaceutical Quality October 29, 2015 (Clarifying Questions Upfront is Key in Process Validation, US and EU PV Principles in Alignment —CDER’s McNally) https://www.ipqpubs.com/2015/10/29/clarifying-questions-upfront-is-key-in-process-validation-us-and-eu-pv-principles-in-alignment-cders-mcnally-stresses/
- EudraLex - Volume 4 - Good Manufacturing Practice (GMP) guidelines - Annex 15 Qualification and validation.
- Pharmaceutical Inspection Convention - Pharmaceutical Inspection Co-Operation Scheme "Guide To Good Manufacturing Practice For Medicinal Products Annexes" July 2018
- European Medicines Agency: Questions and answers on implementation of risk-based prevention of cross-contamination in production and “Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities" 19 April 2018 EMA/CHMP/CVMP/SWP/246844/2018