
5 Keys To Aseptic Processing Improvement & Efficiency
By Hal Baseman, COO and a principal, ValSource, Inc.

Historically, the objective of providing high-quality products and therapies that deliver the best benefit the public could be achieved by assuring that drugs, biologics, and medical devices were manufactured to be safe, effective, and compliant with regulatory requirements. However, that is no longer enough. Today, life science products should also be affordable to patients and a reasonable business proposition for manufactures. These objectives can present challenges to manufacturers as they strive to gain production and process efficiencies.
In 2015, the Parenteral Drug Association (PDA) published its revised Points to Consider for Aseptic Processing (Parts 1 and 2). In those documents, the PDA task force made the case for aseptic processing improvement to keep up with the challenges of modern sterile product manufacturing and distribution. To that end, the document listed five guiding and linked principles for aseptic process improvement and efficiency lie in sterile health care products:
- Science- and risk-based approaches should be used to obtain information needed to make decisions related to the evaluation, design, qualification, operation, and monitoring of sterile product manufacturing processes.
- Technology should be considered to mitigate or reduce the risk to product quality identified in sterile product manufacturing processes and operations.
- Traditional testing and monitoring methods as control strategies should be challenged to ensure they are the best means for aseptic processes.
- New products, therapies, and technologies will present challenges to traditional and existing methods for development, manufacture, validation, and testing of sterile products.
- Global health authority technical and regulatory guidance and requirements should be harmonized with regard to technical language and definitions.1
The following sections will explore each of these principles in further detail, in the context of recent compliance trends.
The Case For Risk-Based Thinking
In recent years, we’ve seen a rise in regulatory concern over adequately determining the risk factors that challenge aseptic and sterile products processing. There needs to be a renewed emphasis on assurance and controls, including leveraging the best available technologies and conducting thorough, science-based risk assessments of processes.2
Quality risk management has become a strong expectation of global health authorities. FDA warning letters increasingly note absence or misuse of risk assessments in regulatory citations. As Table 1 illustrates, there has been a rise in FDA warning letters noting quality risk management-related issues from the introduction of ICH Q9 in 2006 through the first half of 2017.
Table 1: Percent of FDA cGMP-Related Warning Letters with One or More QRM Citations, 2006 to mid-20173
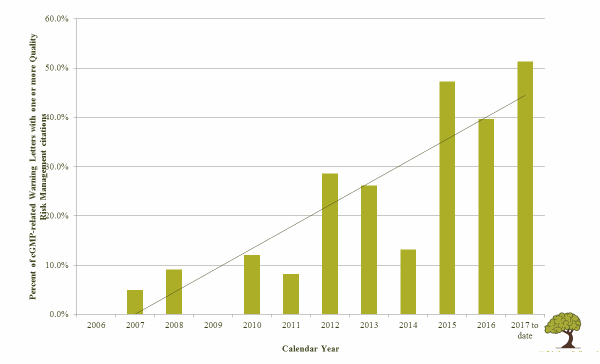
Risk-based thinking is particularly important in aseptic processes, for several reasons. ICH Q9 makes the correlation between risk and uncertainty. The more uncertain something is, the more risk is perceived, and therefore the need for risk management increases.
In aseptic processing, the correlation between what we can observe and the desired outcome (sterility) or the undesired outcome (lack of sterility) is difficult to establish. For example, most would agree that there is a correlation between interventions and disruption of first air with non-sterile products, but we lack the quantifiable data to state with certainty how much disruption will result in failure. Adding to the level of uncertainly is the variation in human behavior and uncertainly of microbiological monitoring and analysis. Simply put, there are things we can observe and monitor that provide indications of aseptic processing performance, but their cause and effect are never completely certain. If you have doubts about this claim, were you ever involved with a media fill you were certain would not pass, but it did? Or did you ever investigate “good” aseptic fills that failed, with no apparent assignable cause?
Where does this leave us? Perhaps we do not know enough about our aseptic processes and have not recognized all the variables that can adversely affect the outcomes. If so, then the best way to control process failure risk is through risk- and science-based process design. It is important to remember that the objective of quality risk management is not merely to identify risk — it is to reduce risk and thus improve the process. Identifying risk, without action, has no material benefit.
The Need For Innovative Technologies
I often begin conference presentations by pointing out to the audience that I started in this industry as an aseptic processing operator nearly 40 years ago. Back then, if I had been asked to predict what aseptic processing might look like 40 years in the future, I doubt I could have envisioned what it is today. And, sadly, that is because it looks pretty much the same as it did back in 1978. Our means of aseptic manufacturing, filling, contamination control, monitoring, and testing have changed little when compared with other technology-driven industries.
The lack of innovation and of technology adoption is increasingly being blamed for ineffectiveness of aging facilities and resultant drug shortages.4 We are still debating the virtue and benefits of isolators. We have yet to embrace continuous process manufacturing or rapid monitoring methods. Our means of detection is relatively unreliable, and our means of manufacturing is still relatively reliant on human performance. Some companies remain reluctant to automate or use electronic record keeping due to concerns over our ability to validate such systems. Regulators are unable to endorse and promote new and innovative means of manufacturing, as they state the need and criticize the lack of technology use. This fear of technology can stifle its efficient use.
I attended a recent trade show where I saw what appeared to be a fully automated aseptic filling process in an isolator. The system used a set of robotic arms to lift and transfer vials from a platform to the filling system. This eliminated much of the risk of fallen vials, and therefore the glove-related interventions needed to remove dropped vials. This, in turn, eliminated the need for gloves at that station and the ensuing risk of pinhole leaks, inadequate decontamination, long vaporized hydrogen peroxide (VHP) cycles, etc. This was clearly a significant process improvement.
As I watched the robotic arms flawlessly execute the operation, I notice a set of round plexiglass ports in the Isolator wall near the operation. When I asked why the ports were there, the representative answered that they were for the glove ports. I asked him why the ports were still needed. He said they weren’t needed, but that customers still insisted upon having them. The incorporation of unnecessary glove ports negated many of the benefits the new technology promised.
All the while, this “new” technology gets older and closer to being outdated. I can still remember the enthusiasm expressed at a 2010 PDA aseptic processing conference when a presenter showed a video of a fully automated sterile product filling line in an isolator. At the end, we thought we had seen a peek into the future. That is, until we were reminded the video had been recorded nearly 10 years earlier. Now, nearly a decade later, how many of these fully automated, operatorless aseptic lines do we see? How long will it take for us to accept newer technologies, such as continuous process manufacturing or closed vial filling?5
Challenging Traditional Approaches
“If it isn’t broken, don’t fix it.”
“We’re not doing so bad.”
“We must be doing something right.”
These three often-heard statements seem to make perfect, practical, common sense. But if we do not feel the need to improve, then we will not improve — and failure to improve cannot possibly be a good business strategy.
In attempting to improve, we certainly should be careful not to over-engineer, over-think, and add redundant control over redundant control to reduce or eliminate the most obscure of risks, with ever decreasing real benefit and value. However, it is important to understand the need to constantly ask the “Why?” and “Why not?” questions when considering how we control aseptic processes. Just because we have done it this way in the past does not always mean it is the best way today.
For example, are full-duration media fills the best way to determine if controls are adequate to address human fatigue and cleanroom environmental conditions? Or would it be more effective to address human factors with more ergonomic process designs, automation, and barrier systems? And wouldn’t it be better to design workflow, barriers, cleanroom HVAC systems, and risk-based environmental monitoring programs to ensure consistent, reliable environments, rather than using “fingers crossed” media fills to demonstrate environmental control? We need to keep our eyes open to process control measures that better link our actions to the establishment and preeminence of product quality attributes — employing true line-of-sight approaches.6
I recall a presentation some years back on the 2011 revision to the FDA Process Validation Guidance.7 The speaker commented that, in the past, an FDA investigator would visit a facility and determine the adequacy of the validation program by confirming that the studies matched written procedures, protocols, and acceptance criteria. The speaker went on to say that the time was approaching when that FDA investigator would visit same the same facility and instead ask: 1) what had the company done to validate the process 2) how it accomplished the objective, and 3) how it would ensure that the process would yield the desired result every time? These are much more difficult question to answer. If an investigator shows up at your facility and asks these questions, how well could you answer them?
Consideration Of New Therapies And Technologies
Will the ways we manufacturing sterile products fit the needs of new therapies and resulting technologies? Many of our standards, guidance, guidelines, advice, and best practices for aseptic processing are based on large-scale manufacturing of replicate doses. The 2004 FDA Aseptic Processing Guidance and the 2008 EU Annex 1 GMPs for Manufacture of Sterile Medicinal Products were written before cell and gene therapy, and other forms of personalized medicines and alternative therapeutic products (ATPs) were introduced.
Autologous products derived from patient apheresis involve different risks and require different manufacturing processes and controls than large-scale, mass-produced medicines. Contamination risks are different. Purification methods are different. Risks associated with identification and chain of custody are different. The timeliness of product release is different. The definition of batches is different. The risk of process failure to the patients may be different.
To complicate the challenges further, many of these products are manufactured using processes that were developed in laboratory or academic settings. Trying to transfer and adapt such processes to the requirements of commercial-scale manufacturing and industry guidance is difficult at best. Can companies fit these processes into existing guidance, or must new guidance be developed? Perhaps the better questions are: Should these products be force-fit into existing technologies, such as laboratory biosafety cabinet manufacturing, or should new and more innovative systems be designed to better fit the product requirements?
A Time For Harmonization Of Regulatory Requirements
We live in an increasingly global business environment. Where the science of aseptic process control agrees, the language and positions of health authorities should (at the very least) be similar. After all, the science and the risk should be the same whether the matter is being considered by U.S., Asian, or European regulators. Having different positions or nomenclature for similar regulatory and technical matters adds the potential for confusion and requires resources to decipher, debate, and address. This detracts from the objective of public health and safety, which each of these global health authorities.
It is encouraging that there are actions and initiatives underway for mutual recognition and harmonization between major global health authorities, including the FDA and EMA. There is hope that the upcoming revisions to the EU Annex 1, prepared by an international Pharmaceutical Inspection Co-operation Scheme (PIC/S) committee, will address some of the remaining differences. However, indications are that some differences may likely remain. In some case these are not differences in position, but in language, as we saw in the respective positions of EU Annex 15 and the FDA Process Validation Guidance on the number of process validation batches proposed.8,9
But in some cases, there may be real differences and conflicts, such as an Annex 1 requirement for pre-use, post sterilization integrity testing (PUPSIT) for product sterilizing filters, verses the need for true science- and risk-based decisions on process design.10
Conclusion
Our objective should always be to modernize and improve aseptic processes. The goal is to provide more efficient manufacturing and assure continued product quality. Improved and better-controlled processes may require the expenditure and allocation of additional resources. There are cost-benefit decisions to be made. However, process quality does not have to come at the expense of manufacturing efficiency and profit. When planned and performed properly, the investment in process understanding and control should result in more reliable processes, higher facility utilization, and less rejected batches, defects, failures, investigations, and regulatory citations.
Careful, thoughtful mapping of the process make it possible to identify and reduce variables and risks. Proper management of risk should result in more effective operations, less rejects and deviations, and higher overall productivity for the manufacturer. The increased understanding of risk and control benefits can also result in greater development and use of innovative technology, through awareness of technology options that meet specific manufacturing needs.
Manufacturers of sterile products must engage in critical thinking, asking “Why?” and “Why not?” questions, challenging and pushing the industry towards more effective means of quality and process control. In this way, we can ensure that we are prepared to meet the challenges of providing sterile pharmaceutical products that are not only safe and effective, but also are available and affordable for the patients who depend on them — and a sound business proposition for the companies that make them.
References:
- Parenteral Drug Association, Points to Consider for Aseptic Processing, Parts 1 & 2, January 2015 and May 2016, Bethesda, MD.
- European Medicines Agency and Pharmaceutical Inspection Co-operation Scheme, “Concept paper on the revision of annex 1 of the guidelines on good manufacturing practice – manufacture of sterile medicinal products”, February 2015.
- Waldron, Greene, and Calnan, “Quality Risk Management: State of the Industry, Part 1. Has Industry Realized the Full Value of ICH Q9?”, Journal of Validation Technology, Vol. 20, No. 4, 2017.
- Woodcock and Wosinska, “Economic and technological drivers of generic sterile injectable drug shortages”, Clinical Pharmacology and Therapeutics, November 2012.
- Parenteral Drug Association, Workshop on Aseptic Processing: Issues and Approaches, November 2010, Bethesda, MD.
- Baseman, Hardiman, Henkels, and Long, “A Line of Sight Approach for Assessing Aseptic Processing Risk: Part I”, PDA Letter, May 31, 2016.
- PDA/FDA Process Validation Guidance Workshop: Meeting Compliance Expectations and Practical Implementation Strategies, April 2011, San Antonio, TX.
- EudraLex, The Rules Governing Medicinal Products in the European Union, Volume 4, EU Guidelines for Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation, March 2015.
- U.S. Food and Drug Administration, Guidance for Industry – Process Validation: General Principles and Practices, January 2011.
- Jornitz et al, “Pre-use/Post-sterilization Integrity Testing of Sterilizing Grade Filters”, PDA Journal of Pharmaceutical Science, September–October 2012.
About The Author:
 Hal Baseman is chief operating officer and a principal at ValSource, LLC and Concordia ValSource, LLC. He has over 39 years of experience in pharmaceutical operations, validation, and regulatory compliance. He has held positions in executive management and technical operations at several drug manufacturing and consulting firms. He is the immediate past chair of the Parenteral Drug Association (PDA) Board of Directors, former chair of the PDA Science Advisory Board, former co-leader of the PDA Process Validation Interest Group, and co-leader of the PDA Aseptic Processing Points to Consider task force, as well as a long-time member of the PDA Training Research Institute faculty. He is a frequent presenter and instructor on subjects related to quality risk management, validation, and aseptic processing. Hal holds an MBA in management from LaSalle University and a B.S. in biology from Ursinus College.
Hal Baseman is chief operating officer and a principal at ValSource, LLC and Concordia ValSource, LLC. He has over 39 years of experience in pharmaceutical operations, validation, and regulatory compliance. He has held positions in executive management and technical operations at several drug manufacturing and consulting firms. He is the immediate past chair of the Parenteral Drug Association (PDA) Board of Directors, former chair of the PDA Science Advisory Board, former co-leader of the PDA Process Validation Interest Group, and co-leader of the PDA Aseptic Processing Points to Consider task force, as well as a long-time member of the PDA Training Research Institute faculty. He is a frequent presenter and instructor on subjects related to quality risk management, validation, and aseptic processing. Hal holds an MBA in management from LaSalle University and a B.S. in biology from Ursinus College.