
Lessons In Navigating The Increasingly Complex World Of Orphan Drug Commercialization
By Ian Love, Ph.D., Senior Business Insights Analyst, Decision Resources Group

Prior to the passage of the Orphan Drug Act of 1983 (ODA), a meager 38 orphan drugs (ODs) were approved, corresponding to — at best — an approved drug for less than 1 percent of an estimated 7,000 orphan diseases. Pooled together, rare diseases afflict a vast number of individuals; the National Organization for Rare Disorders (NORD) estimates 30 million people suffer from one or more rare diseases in the United States alone. Prior to the ODA, market-based solutions to rare diseases were confounded by barriers inherent to the development of niche products. The pharmaceutical industry, already faced with high failure rates and high development costs, likely saw little value in devoting precious capital to developing therapies afflicting small populations.
Since the enactment of the ODA, the FDA has approved over 600 orphan drugs. Applications for orphan drug status have skyrocketed in recent years, and nearly half of all agents approved by the FDA last year had previously received orphan designation (see the FDA’s 2016 Novel Drugs Summary). Clearly, then, the ODA has been an unparalleled success of public policy driving lifesaving innovation, at least in terms of fueling orphan drug approvals. Today, the value of the ODA is evident in the availability of many transformative therapies, including Vertex’s Kalydeco and Biogen’s Spinraza — agents that offer relief to many children suffering from cystic fibrosis or spinal muscular atrophy, respectively. The first of this two-part series will examine rising costs associated with orphan drugs and two related case studies. The second part provides actionable suggestions for developers.
Orphan Drug Innovation Accounts for Increasing Proportion of Payer Budgets
The ODA has been wildly successful in its primary goal of spurring innovation that has led to lifesaving therapies. Unfortunately, the design of the ODA and several of the incentives the act provides have led to a host of unintended consequences, some of which are proving to be costly — potentially unsustainably — to taxpayers, policy holders, and patients. Indeed, a survey of U.S. payers conducted by Decision Resources Group (DRG) in 2016 indicated 16 percent of surveyed managed care organizations’ (MCOs’) annual drug spend was allocated to ODs, and the same payers expected that figure to rise to 21 percent in three to five years (Figure 1); in contrast, surveyed payers do not expect the percentage of annual drug spend allocated to non-rare specialty drugs to increase over the same time frame (Figure 2). Furthermore, orphan drug tax credits alone (arguably one of the least costly provisions of the ODA) are estimated by the U.S. Treasury Department to exceed $9 billion annually by 2025 — up from an already substantial estimated expenditure of $2 billion in 2017 (U.S. Treasury Department Office of Tax Analysis, November 11, 2015) — representing an astonishing CAGR in excess of 16 percent.
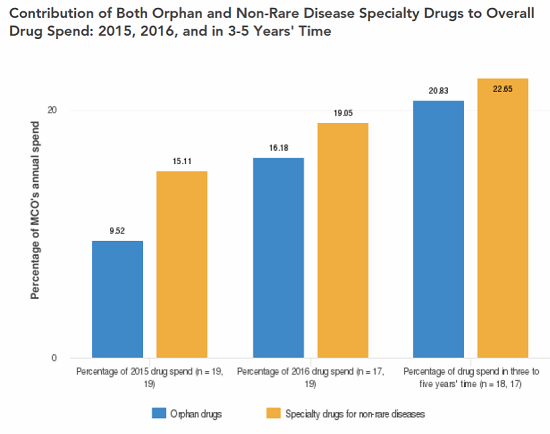
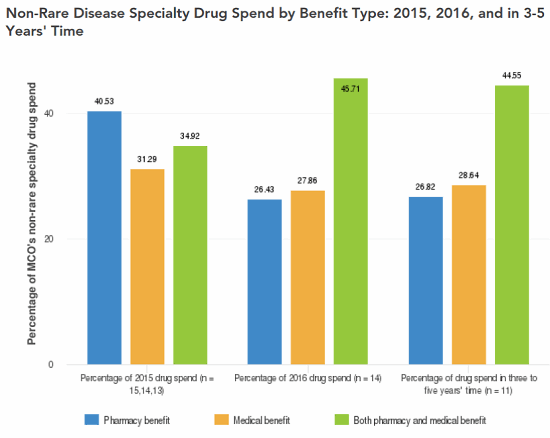
Several key issues underlie the growth in healthcare costs as a result of the ODA. Chief among them are:
- Some such agents become multi-blockbuster medications by obtaining orphan drug designation (ODD) yet eventually gain approval for additional, sometimes non-orphan, indications, thereby garnering exclusivity that may help to protect sales across all approved indications. Roche’s Rituxan, for example, first received marketing approval in the U.S. in 1997 for the treatment of relapsed or refractory CD20-positive B-cell non-Hodgkin’s lymphoma, for which Roche obtained ODD. It reached U.S. sales in excess of $1 billion in 2002 and has since attained numerous additional orphan and non-orphan indications. One challenge inherent to the definition of what, precisely, should be considered an orphan disease is that as our understanding of disease pathophysiology grows, it often becomes evident that drivers of more prevalent indications are also at work in certain rare diseases. Such fortuitous advances offer developers the opportunity to obtain more and more orphan designations which each confer the benefits laid out in the ODA — most notably, seven years of market exclusivity regardless of the status of the patent. Furthermore, even if payers renegotiate discounts upon label expansions, total drug costs continue to rise.
- The particularly high price tag of many orphan drugs is another key driver of growing pharmacy cost. The median price of an orphan drug currently stands at $100,000 per patient per year, and several command prices in the $300,000 to $400,000 range. Assuming a small rebate of about 10 percent, an orphan drug priced at $350,000 would reach blockbuster status by treating just over 3,000 patients.
With the number of annual orphan drug approvals in the range of 30 to 40 each year, high prices and expanding eligible patient populations will inevitably strain budgets. Indeed, many orphan drugs set to launch within the next few years are expected to rapidly reach blockbuster status even without further label expansions; DRG research indicates Spinraza and the gene therapy AVXS-101, if launched, will garner sales of $1 billion by 2025 in the U.S. alone (2016 Spinal Muscular Atrophy Niche & Rare Disease Landscape & Forecast; Decision Resources Group). Further, seven years of market exclusivity granted by the ODA limits payers’ leverage when negotiating pricing with developers, particularly when no other approved therapies exist.
In the absence of broadly accepted cost-effectiveness measures in the U.S., developers must approach payers, who increasingly recognize the growing burden of OD costs, with independent, potentially inconsistent, pricing strategies. Some of these strategies could negatively impact formulary status or tiering among payers’ commercial plans and ultimately may cost developers substantial revenue. Although the conversation is ongoing, standardized methods of cost-effectiveness analysis appropriate for the rare disease space simply don’t exist.
Spinraza: An ODA Success Story
Biogen/Ionis’s antisense oligonucleotide nusinersen (Spinraza) exemplifies how the ODA is capable of promoting the development of medications for difficult-to-treat rare diseases. Spinraza reached the market in early 2017 as the first approved therapy for spinal muscular atrophy (SMA). SMA is a severe pediatric disease that has been otherwise lacking in effective treatment options. Based on clinical criteria alone, the estimated treatable population of Spinraza stands at fewer than 4,000 patients. Spinraza performed admirably in a well-designed and powered placebo-controlled trial in the most severe (Type I) SMA patients, as well as in open-label trials with less severe (Types II and III) patients. Interim analyses of a pivotal Phase III placebo-controlled trial in Type I patients indicated patients receiving Spinraza achieved a greater number of motor milestones than patients receiving a sham procedure (Biogen press release, August 1, 2016). Furthermore, over half of the SMA patients who received Spinraza were alive after one year, in contrast to only 32 percent of patients who received a sham treatment.
Biogen announced the price of Spinraza would be $125,000 per intrathecal injection — six injections in the first year, equating to $750,000, and three each year thereafter, or $375,000 — a price point that may not be supported by conventional cost-effectiveness analyses used to inform pricing and reimbursement decisions in the pharma industry. Although Spinraza benefits from an exemplary clinical profile, not every orphan drug will bring as compelling a data package, even if the potential clinical gains are meaningful (i.e., small population precludes statistical power in a trial). Thus, it is critical that orphan drug developers recognize the opportunity to influence policy in this arena through early engagement with payers and other stakeholders.
Emflaza: The Challenges Surrounding the Assessment of Orphan Drug Value
The recent FDA approval of Emflaza (deflazacort) for Duchenne muscular dystrophy (DMD) highlights precisely why the orphan drug pricing process needs the development of a cost-effectiveness framework defined by payers, policy makers, and developers. Developers need to interface with payers and other entities regulating market access. Deflazacort was previously affordable and accessible, albeit requiring importation, to many patients in the United States. The drug is one of several glucocorticosteroids used to treat patients with DMD. The agents help to preserve muscle function and strength and are widely generically available. While prednisone and prednisolone are generally used to treat DMD patients, concerns over side effects such as weight gain often lead patients or physicians to alternatives. Limited clinical data suggests deflazacort promotes less weight gain than prednisone in DMD patients1; however, the magnitude and long-term clinical importance of the difference remains in question.
Marathon Pharma undertook development of deflazacort for DMD in the United States and ultimately earned regulatory approval for the drug, under the brand name Emflaza, in early 2017 — thereby alleviating the need for importation of a potentially beneficial medication for DMD patients. However, a swift public outcry followed Marathon’s pricing announcement — a list price of $89,000/year — which led the company to halt the agent’s launch and ultimately sell the asset. Notably, data from a survey of payers in the United States conducted by DRG in 2016 suggests receptivity to Emflaza would be limited even at annual price points well below Marathon’s intended price. Approximately one-third of surveyed MCOs indicated they would cover deflazacort if the agent were priced at $30,000 per patient per year; it achieved preferred status among more than half of surveyed MCOs when priced at or under $15,000 per patient per year.
Thus, what began as a pricing misstep has likely delayed the market entry of a potentially useful medication for DMD patients — one that could potentially have been avoided with an early approach to payers to frame a discussion around the medication’s true value.
In Part 2, we provide recommendations to developers of orphan drugs on how they can increase access to their medications and influence policy in this evolving space.
References
- Griggs RC, et al. “Efficacy and safety of deflazacort vs. prednisone and placebo for Duchenne muscular dystrophy.” Neurology. 2016; 87(20):2123-2131.
About The Author:
 Ian Love, Ph.D., is a senior business insights analyst in the Infectious, Niche, and Rare Diseases department at Decision Resources Group. He specializes in atopic dermatitis and a diverse group of rare diseases. He received his doctorate in biomedical sciences from the University of Massachusetts Medical School and his B.S. in cell and molecular biology from Worcester Polytechnic Institute. Prior to joining DRG, Love was an instructor in the Department of Internal Medicine at Virginia Commonwealth University, where his work focused on understanding the regulation of programmed cell death by the tumor suppressor p53.
Ian Love, Ph.D., is a senior business insights analyst in the Infectious, Niche, and Rare Diseases department at Decision Resources Group. He specializes in atopic dermatitis and a diverse group of rare diseases. He received his doctorate in biomedical sciences from the University of Massachusetts Medical School and his B.S. in cell and molecular biology from Worcester Polytechnic Institute. Prior to joining DRG, Love was an instructor in the Department of Internal Medicine at Virginia Commonwealth University, where his work focused on understanding the regulation of programmed cell death by the tumor suppressor p53.