
Managing Organic Impurities And Nitrosamines In APIs
By Stefan Kettelhoit, Ph.D., and Norbert Waldöfner, Ph.D.

Active substance impurities as a whole – and therefore also organic impurities in particular – can have a variety of causes. Based on the effect organic substances have on genetic material in experimental in vitro tests, certain substances can be identified as genotoxic (the effect is referred to as genotoxicity). This effect is part of the general toxicity assessment of substances.
In this context, the terms mutagenicity and carcinogenicity are used if, in addition to in vitro genotoxicity, a mutagenic (gene-altering) carcinogenic (cancer-promoting) or teratogenic (embryo malformation-inducing) effect has been proved or suspected in vivo.
Mutagenic Substances
The ICH guideline M7(R2) on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk, which was implemented in the European Union as the guideline EMA/CHMP/ICH/83812/2013, is authoritative for the assessment and control of mutagenic substances in active substances.
This guideline should be regarded as a supplement to the following guidelines:
- ICH Q3A (R2) Impurities in new drug substances
- ICH Q3B (R2) Impurities in new drug products
- ICH M3 (R2) Nonclinical safety studies for the conduct of human clinical trials and marketing authorisations for pharmaceuticals.
The M7 guideline provides methods for establishing limits for mutagenic impurities below which only a negligible risk of cancer is expected. In addition, measures for assessing and controlling these mutagenic impurities are presented.
The M7 guideline advocates using the threshold of toxicological concern (TTC) for controlling mutagenic substances. This defines acceptable intake (AI) values that are associated with a negligible cancer risk. It is assumed that an intake of 1.5 μg/day of a mutagenic impurity is associated with an acceptable risk for the person concerned.
All impurities that may arise during the synthesis and storage of an active substance (potential impurities) must be evaluated. This includes evaluating both previously identified impurities and potential impurities that may arise during the synthesis of the active ingredient or because of subsequent degradation processes.
The risk of these impurities being carried over into the active substance must be assessed in relation to the synthesis route. This also applies to the use of contaminated active substance starting materials or intermediates.
A risk-based assessment of impurities in active substances regarding their genotoxic or mutagenic potential may also depend on the synthesis step. It is therefore recognized that, for example, the carryover of impurities from early synthesis steps may be disregarded if a risk-based justification is available.
Impurities and degradation products formed during synthesis, which could ultimately also be contained in the active substance, must be assessed with regard to their genotoxic or mutagenic potential using the methods specified in Section 6 of the M7 guideline.
A suitable control strategy may include, for example, the following aspects in addition to analytical testing of active substances and medicinal products:
- material attributes of raw materials, starting materials, intermediates, reagents, solvents, and primary packaging materials
- facility and equipment operating conditions
- the development of the manufacturing process.
In-depth knowledge of the chemical processes involved in active substance production and of the stability of the active substance is of paramount importance. A control strategy must also take into account the principles of quality risk management.
Nitrosamines
Nitrosamines (more precisely, N-nitrosamines) attracted worldwide attention in 2018 when they were discovered to be present as unexpected impurities in various active substances. Initially, antihypertensive agents from the sartan group were affected. The discovery of these impurities led to product recalls worldwide.
The EMA felt compelled to issue guidelines on preventing nitrosamines in medicinal products. This has resulted in far-reaching obligations for marketing authorization holders. Among other things, they are required to check their products for risks associated with the presence of nitrosamines or the potential for them to be introduced. This involves identifying all active substances that could potentially be contaminated with N-nitrosamines. Analyses must be carried out on the relevant products and the results reported to the EMA.
The EMA has made the relevant regulatory processes and templates available on its website. The EMA's central website “Nitrosamine impurities” contains links to other relevant documents. Of particular note is document EMA/526934/2019, titled Lessons learnt from presence of N-nitrosamine impurities in sartan medicines. This document contains detailed further considerations, including a revision of important international guidelines as well as audits and inspections of active ingredient manufacturers.
From their own experience, the authors can report that the topic of nitrosamines has now become an established part of GMP audits. Active ingredient manufacturers have generally carried out corresponding risk analyses for all active ingredients manufactured and can refer to specific documents and templates for this purpose.
The Assessment Report EMA/369136/2020, published in June 2020 and titled Nitrosamine impurities in human medicinal products, provides a detailed summary of the current state of knowledge on nitrosamines in human medicinal products.
As regulatory developments in the field of nitrosamines are very dynamic, it is essential to research the currently applicable requirements in a timely manner. In the EU, the EMA website and the Q&A document are particularly noteworthy as these two sources of information are updated frequently. Extensive updates were made to the Q&A document in 2023. The responsibility of the marketing authorization holder with regard to controlling nitrosamine impurities throughout the entire life cycle of a medicinal product is referenced.
The Q&A document includes approaches for categorizing N-nitrosamines in terms of their carcinogenic potential. Three appendices are particularly noteworthy:
- Appendix 1 lists the N-nitrosamines for which AIs have already been established.
- Appendix 2 introduces the Carcinogenic Potency Categorization Approach (CPCA).
- Appendix 3 describes extended Ames test conditions for N-nitrosamines.
The EDQM has defined measures for CEP holders, which it has summarized in its newsroom.
The guidance published by the FDA in September 2020 (Control of Nitrosamine Impurities in Human Drugs) provides valuable advice on how to avoid nitrosamine impurities. Another FDA guideline worth mentioning is Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs) from August 2023.
Figure 1: Key documents and internet links on nitrosamine impurities in human medicines
The European Pharmacopoeia addresses the topic of N-nitrosamines in several monographs/texts:
- Monograph 2034 (substances for pharmaceutical use)
- Monograph 2619 (pharmaceutical preparations)
- Chapter 2.5.42 (N-nitrosamines in active substances)
The monographs set out the requirements relevant to manufacturers of active substances and medicinal products. Affected manufacturers should minimize risk as far as possible by, for example, adapting the manufacturing process. In addition, they should implement a control strategy to enable the detection and control of N-nitrosamine impurities.
What Are Nitrosamines?
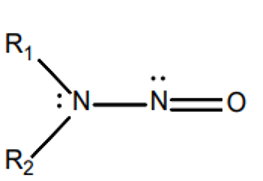
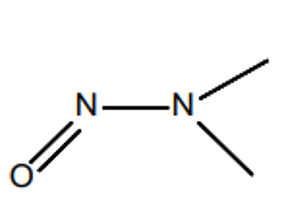
N-nitrosamines are a class of substances with the general structural formula R1R2N-NO, where R1 and R2 are alkyl or aryl radicals. N-nitrosamines belong to the N-nitroso compounds. Based on animal studies, nitrosamines are classified as potentially carcinogenic. According to ICH M7(R2), nitrosamines belong to the group of highly potent mutagenic carcinogens referred to as a “cohort of concern.” The best-known representatives are NDMA (N-nitrosodimethylamine) and NDEA (N-nitrosodiethylamine).
 |
 |
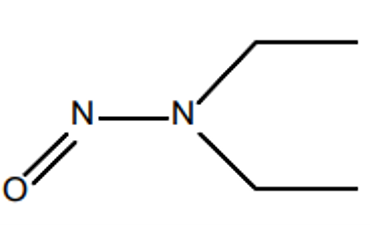 |
| General structural formula | NDMA | NDEA |
Figure 2: Chemical structural formulas of nitrosamines and two examples (NDMA (N-nitrosodimethylamine) and NDEA (N-nitrosodiethylamine))
Put simply, the mutagenic effect of nitrosamines is based on their metabolic activation, which produces alkyl or aryl diazonium ions.
Based on the recommendations of ICH M7 and various assumptions, preliminary substance-specific AI levels for various N-nitrosamines were published in 2019. Taking into account the maximum daily doses, maximum concentration limits for impurities in active substances were derived from these.
Meanwhile, both the EMA and the FDA have published acceptable intake levels for various N-nitrosamines and specified the methods for determining AIs.
How Should Limits For N-Nitrosamines Be Set?
As previously mentioned, N-nitrosamines are considered substances that belong to a cohort of concern according to ICH M7(R2). For these substances, the TTC of 1.5 µg/day cannot be routinely applied. Therefore, AIs must be established on a substance-specific basis, for which a negligible risk can be expected.
The calculation of AIs is based on the maximum daily dose of the medicinal product being administered for a lifetime. According to the EMA Q&A document, the less than lifetime (LTL) approach should only be considered as a temporary measure, after consultation with the competent authorities.
Based on the AIs, limit values for medicinal products can be determined.
How Are N-Nitrosamines Formed?
Q&A 4 of the EMA Q&A document sets out the risk factors relating to the manufacture of active substances in detail.
In general, N-nitrosamines can form when certain amines and a nitrosating agent are combined under specific conditions. The EMA Q&A document also mentions other possible formation pathways, such as oxidation and reduction processes from hydrazine-like compounds and N-nitro derivatives.
The main reason for the formation of nitrosamines during chemical synthesis of active substances is the presence of nitrite salts and esters or other nitrosating agents in the presence of secondary and tertiary amines.
Examples of nitrosating agents are:
- sodium nitrite,
- alkyl nitrites,
- nitrosohalides,
- nitrosonium salts,
- nitrogen oxides,
- nitroalkanes,
- halogenated nitroalkanes,
- potassium nitrosodisulphonate, and
- nitroso sulfonamides.
Sources of secondary, tertiary, or quaternary amines in active ingredient synthesis can include:
- the active ingredient itself,
- intermediates,
- starting materials,
- raw materials and reagents,
- catalysts, or
- solvents.
The amine functionalities may be a component of these substances or may be present as impurities. They may also arise through the degradation of certain substances.
In general, certain technical or GMP aspects of active ingredient production can also lead to increased risks:
- Use of multipurpose equipment
- Inefficient manufacturing instructions or cleaning instructions that lead to the carryover of impurities between process steps
- Use of contaminated recycled or reused materials
In the authors' view, using contaminated raw materials ordered from suppliers is a risk that is often overlooked. Such materials may contain residual nitrosamines or nitrosating agents. This makes careful supplier qualification even more important.
Other specific risks in active substance manufacturing mentioned in the EMA Q&A document are:
- nitrite formation through the oxidation of hydroxylamine, or nitrite release from nitroaromatic precursors
- use of disinfected water in the presence of secondary or tertiary amines
- oxidation of hydrazines, hydrazides, and hydrazones
- carryover of nitrosamines intentionally generated during the manufacturing process.
How Are Nitrosamines Determined?
Normally, the exact determination of the concentration of N-nitrosamines in active substances is naturally of particular importance. It is the responsibility of the marketing authorization holder to develop a suitable analytical method with the required sensitivity.
The pharmacopoeia monographs refer to Chapter 2.5.42 of the European Pharmacopoeia for the determination of N-nitrosamines, which lists three analytical methods:
- Method A: LC-MS/MS
- Method B: GC-MS
- Method C: GC-MS/MS
The aforementioned methods are considered validated for certain combinations of N-nitrosamines and active substances. For other combinations, the methods must be validated.
In 2018, the official medicinal product control laboratories (OMCLs) of the General European OMCL Network (GEON) began developing methods for determining N-nitrosamines. These methods were subsequently published on the EDQM website for informational purposes.
How Can The Formation Of Nitrosamines Be Prevented?
Cross-contamination or errors caused by production personnel during the production of active substances can cause nitrosamine formation. To prevent N-nitrosamine formation during the manufacture of active substances, the aforementioned reaction conditions should be avoided, by, for example, using alternative solvents or reagents where possible.
Additionally, if there is a risk of contamination by nitrosamines, the raw materials used should be subjected to intensive evaluation and testing. Suppliers of raw materials for active ingredient production should also be qualified.
The use of recovered solvents should be carefully evaluated, or it should be clearly demonstrated that any nitrosamines contained in the solvents can be completely removed by purification processes. Commissioning external companies for solvent recovery should also be reviewed.
Regarding solvent recovery, the following scenarios are recommended:
- Use solvents recovered from the same process step in the synthesis of the active substance.
- Carry out solvent recovery internally.
- Use dedicated equipment for solvent recovery.
- Evaluate and, if necessary, validate cleaning procedures for the equipment used for recovery.
- Carefully document cleaning steps and check them in accordance with the dual control principle.
Further necessary measures depend on the specific results of the respective root cause analysis and must be initiated considering the current state of knowledge and the principles of quality risk management.
Editor's Note: This article is a shortened excerpt from GMP knowledge contained in the online portal GMP Compliance Adviser, which provides in-depth information about GMP best practices and regulations with a focus on Europe but also referring to the U.S., Japan, and others (PIC/S, ICH, WHO, etc.).
About The Authors:
 Stefan Kettelhoit, Ph.D., is a pharmacist and freelance consultant for life sciences companies. He has many years of experience in the areas of development, pharmaceutical technology, and international project management.
Stefan Kettelhoit, Ph.D., is a pharmacist and freelance consultant for life sciences companies. He has many years of experience in the areas of development, pharmaceutical technology, and international project management.
 Norbert Waldöfner, Ph.D., has been working for an ISO 17020 Type A-accredited GMP audit service provider since 2011. He has audited more than 200 companies worldwide, including manufacturers of starting materials and active ingredients for pharmaceutical products, as well as other suppliers and service providers.
Norbert Waldöfner, Ph.D., has been working for an ISO 17020 Type A-accredited GMP audit service provider since 2011. He has audited more than 200 companies worldwide, including manufacturers of starting materials and active ingredients for pharmaceutical products, as well as other suppliers and service providers.