
Managing QMS Disparities Between Drugs And Devices For Combination Products
By Bikash Chatterjee, CEO, Pharmatech Associates
 Combination products represent a remarkable opportunity to extend the potential patient population for biologics and improve the delivery of drug therapies. This is one of the most dynamic segments in the life sciences , projected to grow to $115 billion by 2019.1 A combination product is defined as a product composed of any combination of a drug and a device. This could be a biological product and a device; a drug and a biological product; a drug and a device; or a drug, device, and biological product. Each component of a combination product is regulated by different chapters of the Code of Federal Regulations (CFR), which adds a layer of complexity for many organizations developing them.
Combination products represent a remarkable opportunity to extend the potential patient population for biologics and improve the delivery of drug therapies. This is one of the most dynamic segments in the life sciences , projected to grow to $115 billion by 2019.1 A combination product is defined as a product composed of any combination of a drug and a device. This could be a biological product and a device; a drug and a biological product; a drug and a device; or a drug, device, and biological product. Each component of a combination product is regulated by different chapters of the Code of Federal Regulations (CFR), which adds a layer of complexity for many organizations developing them.
Here is the regulatory dilemma: Drugs are regulated by 21 CFR 210/211, medical devices by 21 CFR 820, HCT/Ps (human cells, tissues, or cellular or tissue-based products) by 21 CFR 1271, etc. An organization’s ability to harmonize its product development and quality management systems (QMS) to accommodate these differences is central to the successful commercialization of a combination product.
In the first of this two-part series, we will examine some of the common pitfalls drug companies face when they pursue the development of a combination product.
Regulations and Guidance
In 2013 the FDA released 21 CFR 4, which greatly clarified which elements of all applicable regulations must be included for drug-device single entities and co-packaged combo products — and provided a more streamlined path forward for combination devices. The rule states that companies pursuing a combination product need only adopt specific sections of the corresponding CFR. In other words, a drug company that relies on CFR 210 and 211 has the option of fully adopting both regulations, or only adopting specific sections of CFR 820, and vice-versa for device companies. The alternatives are illustrated in Figure 1 below.
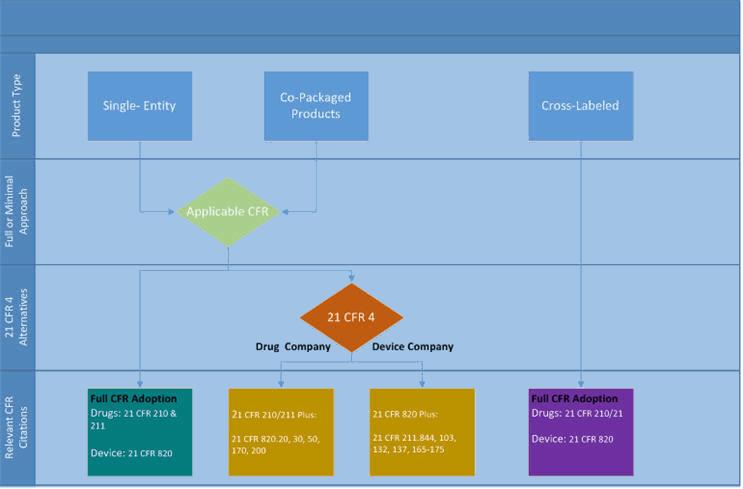
Figure 1: Applicable CFR regulations for combination products as defined by 21 CFR 4
Still, the challenge for organizations pursuing a positive combination product submission lies in translating these requirements into practice. If an organization can anticipate the intersection between these systems, then it is possible to move smoothly through the development process to foresee and fulfill the FDA requirements.
Drug and Device Development: What’s the Difference?
The nice thing about drugs and devices today is that they are more similar in terms of their development activities than ever before. The implementation and enforcement of the 2011 Process Validation Guidance in the U.S. has pushed the pharma industry to pursue a more structured development process incorporating components of Quality by Design (QbD). Regulated devices in the U.S. must follow well-defined design control procedures and be developed using the appropriately defined consensus standards adopted by the FDA according to complexity, risk, and regulatory strategy.
One perspective when integrating CFR 820 with 210/211 QMS is to look for the overlaps between the two frameworks. As far as product and process development is concerned, both systems incorporate a verification and validation framework. However, when looking closer there are differences in the degree and timing of these activities. Utilizing 21 CFR 4 as a guideline, it is possible to highlight any key intersections between the two quality frameworks that can cause difficulty for an organization as the product moves through development. To illustrate, we will consider the potential challenges for a drug company that is developing both a new chemical entity and a medical delivery device as part of a new drug application (NDA).
Quality Management Systems (QMS)
If a drug manufacturer is responsible for device development and manufacturing, it is important to bridge the differences between the compliance regulations. Regulated devices are governed by CFR 820 — also called the Quality System Regulation (QSR). However, device suppliers are likely to follow ISO standards, such as ISO 13485, ISO 9001, EN 46001, or ISO 15378. None of these standards are identical to the QSR, and they differ from the drug GMP requirements defined by CFR 210/211 (or CFR 606-680 for biologics).
The differences can be significant depending upon system requirements. For example, ISO 13485 does not specifically mandate that controls be in place for manufacturing materials, although the expectation is that they will be. These two elements are emphasized in both 210/211 and 820. CFR regulations are also required by law, while guidances describe the agency’s expectations but are not enforceable as law. Understanding the differences in each standard is important to calibrating the level of QMS required at suppliers, sub-suppliers, and intermediate processors. It is an important consideration when developing the supplier qualification program.
Design Control and Specification Setting
Devices must follow a design control process and that means creating a process that results in a complete design history file (DHF). The 10 key elements of a design controls process are listed below: Their relationship to the design control process is shown in Figure 2.
- Design planning
- Design inputs
- Design outputs
- Design reviews
- Risk management
- Design verification
- Design validation
- Design transfer
- Design changes
- Design history file
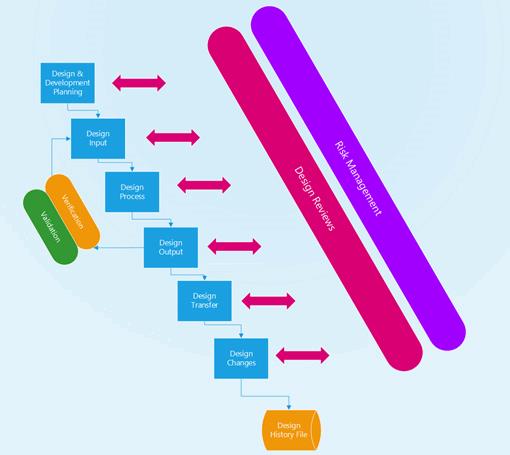
Figure 2: Medical device design control process
Design controls should begin as the product is moving from the concept phase to the feasibility phase. The rigor around this element must be very high. If the device application is being filed as part of the NDA or BLA (biologics license application) without separate approval via FDA’s Center for Devices and Radiological Health (CDRH), regulatory authorities will expect a very high level of control, especially if software is a component of the delivery system. The FDA’s newly created Office of Combination Products (OCP) will designate an FDA center to have primary jurisdiction over the submission for combination and non-combination products. The goal of the center is to stabilize the premarket reviews of all submissions.
Within pharma QMS, oversight a number of typical pitfalls exist. Here are four:
1. Design planning: Today, most drug product and process design starts with a quality target product profile (QTPP). This document captures all the salient product design considerations including performance, container, and product critical quality attributes (CQA). Device design planning does not utilize this framework, but for pharma companies it is a reasonable way to flag and frame the primary elements of the system’s performance at the beginning of the design process. An example of a QTPP for a small molecule parenteral coupled with a prefilled syringe is shown in Table 1. Note the system delivery performance component of the QTPP.
Table 1: Combination Product QTPP Example
|
Therapeutic indication: |
Pain management |
|
Clinical setting: |
Use in home |
|
Route of administration: |
Subcutaneous injection |
|
Dosage strength: |
4 mg and 6 mg |
|
Pharmaceutical form: |
Sterile liquid |
|
Container closure system: |
Glass prefilled syringe with elastomeric plunger in spring-loaded autoinjector |
|
Drug/Device Product Quality Criteria |
|
|
Shelf-life: |
Not less than (NLT) 2 years when stored at 2 to 8°C |
|
Degradation products: |
Meets criteria of ICH Q3B |
|
Sterility: |
Meets USP <71> |
|
Endotoxin: |
Meets USP <85> |
|
System delivery performance: |
Injection depth: suitable for subcutaneous injection Injection volume: suitable to deliver required dose Activation force: minimum to prevent accidental actuation; maximum not to exceed customer need Injection time: target less than 5 seconds to deliver required dose |
2. Poor Configuration Management: This is a common issue during the design review, verification, and validation process. This speaks specifically to what version of software/device and configuration of the device was used in the review and for testing. It is essential to have full traceability to the systems being used in development and testing, to be able to link the design decisions and testing results to the final system performance.
3. Risk Analysis: ISO 14971 requires hazard analysis and design, process, and user failure mode effects analysis (FMEA) to ensure that these structures are formalized and appropriately constructed (e.g., at least four risk rankings). Pharma does not mandate risk assessments but rather relies on ICH Q9 and Q10 as reference models for industry best practice. The context of each FMEA is very specific, especially the user FMEA intended to understand the potential system harm. Often this cannot be determined without an physician or similar clinical expert who can evaluate the true potential for patient harm.
Conclusion
The design control process represents a significant departure from typical drug development. The rigor required for the design and development of products is very well defined. As the device and drug development converge, it is essential for the R&D and QMS to clearly specify the inputs and outputs that will ensure the traceability, and for checks and balances to be created.
In Part 2, we will discuss some of the unique challenges associated with product quality measurement, product release, and clinical device management.
References
1. Global Drug Device Combination Products Market to Surge at 7.9% CAGR 2013-2019 as Hospitals Adopt Minimally Invasive Surgeries, Transparency Research http://www.transparencymarketresearch.com/pressrelease/drug-device-combination-market.htm
About The Author
Bikash Chatterjee is president and chief science officer for Pharmatech Associates. He has over 30 years’ experience in the design and development of pharmaceutical, biotech, medical device, and IVD products. His work has guided the successful approval and commercialization of over a dozen new products in the U.S. and Europe.
Mr. Chatterjee is a member of the USP National Advisory Board, and is the past-chairman of the Golden Gate Chapter of the American Society of Quality. He is the author of Applying Lean Six Sigma in the Pharmaceutical Industry (ISBN: 978-0-566-09204-6) and is a keynote speaker at international conferences. Mr. Chatterjee holds a B.A. in biochemistry and a B.S. in chemical engineering from the University of California at San Diego.