
Nitroso Impurities In Valsartan: How Did We Miss Them?
By Ketan Agravat, OMSAV Pharma Research

On July 17, 2018 and July 18, 2018, respectively, the EMA and the FDA reported a major issue regarding the detection of a genotoxic impurity, NDMA (N-nitrosodimethylamine), and subsequently NDEA (N-nitrosodiethylamine), in current lots of valsartan,1 an API used to manufacture generic angiotensin receptor blockers (ARBs). The API at issue was supplied by two Chinese companies, Zhejiang Huahai and Zhejiang Tianyu, and was later discovered in API manufactured by Indian manufacturer Hetero Labs. Both NDMA and NDEA are considered genotoxic compounds as they contain a nitroso group, which is a gene mutating group. This concern has spread across the EU, Canada, and the U.S., and is expanding to other regions as well.
NDMA is found in nature and food, including in some water supplies, in very low levels.2 Consuming up to 96 nanograms NDMA/day is considered reasonably safe for human ingestion.3
Probable Origin Of NDMA/NDEA And Its Impact On Other APIs
Most ARBs have a chemical structure that includes a tetrazole group. It appears that tetrazole ring formations, coupled with certain manufacturing conditions using the solvent N, N-Dimethylformamide, aka DMF, gave rise to this class of impurities in drug substance intermediates used in sartans. Other tetrazole ring formations make up APIs like candesartan, losartan, irbesartan, and olmesartan. The impurity may also roll over to other compounds normally known as “zoles,” such as omeprazole, etc.4,5,6
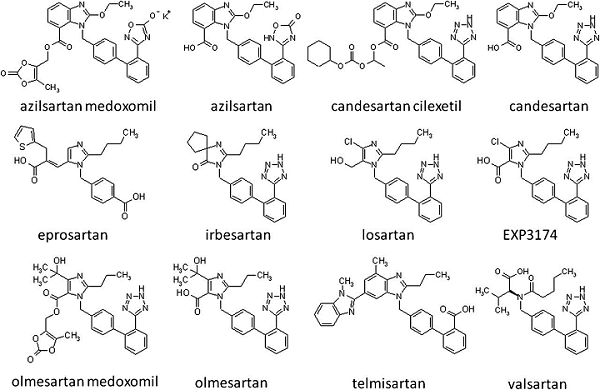
Figure 1: Structure of sartans
The nitroso impurity may well roll over to other tetrazole rings containing such molecules as cefonacid, ceforanide, cefoperazone, cefmetazole, colostazole, latamoxef, parnlukast, and pemiroplast, to name a few.
Regulatory Assessment Of The Process Change
It is not known whether the solvent was newly introduced into the manufacturing process during the process changes that occurred in 2011-2012. However, as reported by various agencies, these impurities started to appear in the product after the change in the process; hence, it is likely the solvent was newly introduced in the synthesis, and the following assessment is based on that assumption. On the contrary, however, Huahai U.S. has issued a statement indicating that the impurity appears only in recent lots.7
European Perspective
According to the-then-official European Directorate for the Quality of Medicines (EDQM) guideline for revisions to an approved certificate of suitability (CEP)8 at the time of the change submitted by the manufacturer, “MAJ3 Substantial change to the manufacturing process/addition of an alternative manufacturing process for a starting material, intermediate or final substance likely to change the qualitative and/or quantitative impurity profile (e.g., new reagents, solvents, materials are introduced in the synthesis).”
This clearly indicates that when a new solvent is introduced, it is considered a major change and will be assessed accordingly by the respective agency in line with the requirements for assessment of genotoxic impurities. However, the manufacturer missed reporting the generation of the new impurity (NDMA/NDEA, genotoxic impurity) in the revised process.
A revised EDQM guideline for CEP revisions, shown in Table 1, was published in July 2014.9
Table 1: Assessment of Revision in CEP Application
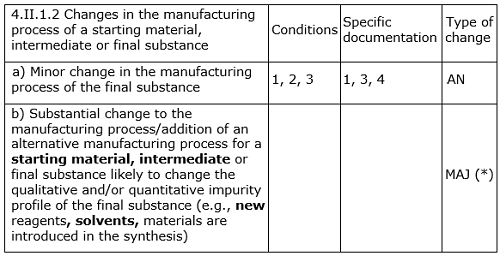
It is again very clearly indicated in this and other relevant guidelines that if the proposed change has significant impact on the qualitative and/or quantitative impurity profile, then the levels of known, unknown, and total impurities found in the post-change batches should be assessed/compared critically with respect to with those of pre-change batches.
EDQM subsequently (September 2018, with implementation date of January 2019) changed10 the above revision guideline to include evaluation for genotoxic impurities as well.
Interestingly, the manufacturer, Zhejiang Huahai, stated that the change was implemented in 2012 after approval by relevant regulatory authorities,11 without clarifying which authorities approved the process change.
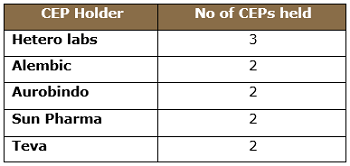
Approximately 28 CEPs for valsartan are approved by EDQM, as tabulated below, with various statuses found in the EDQM CEP database. Five companies hold more than one CEP, meaning they are using more than one manufacturing process.
Table 2: Major Valsartan CEP Holders
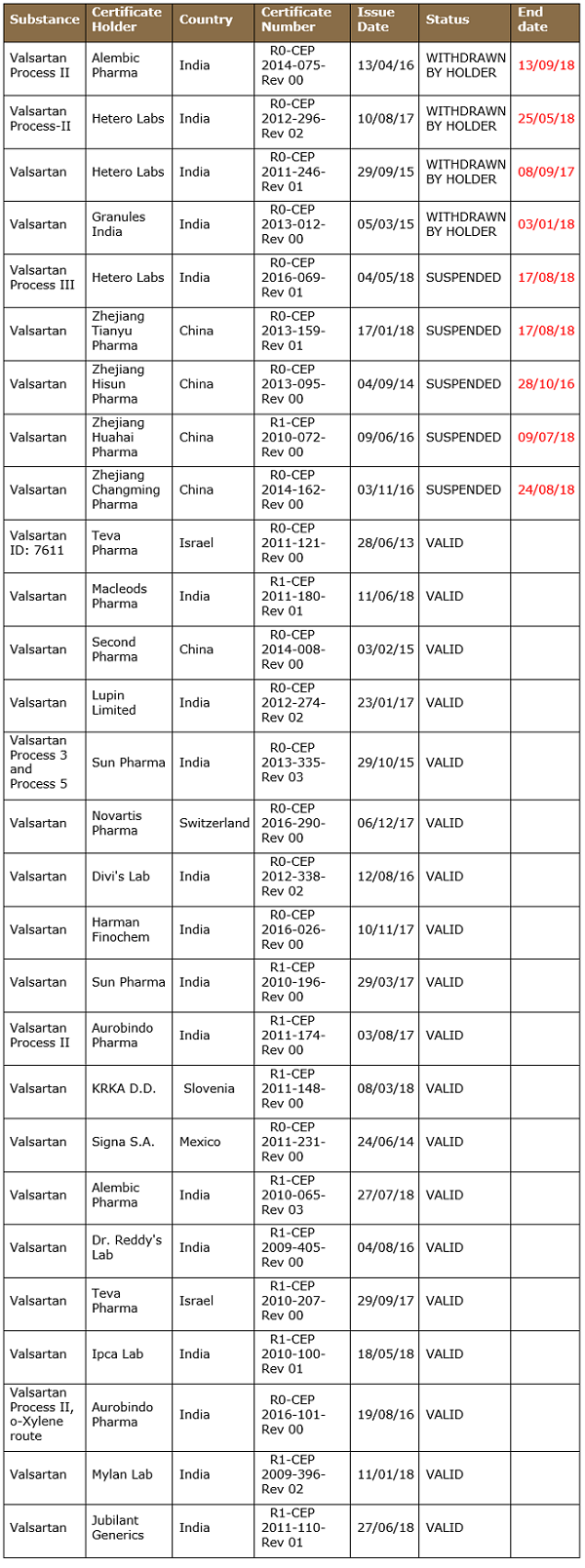
EDQM has suspended four CEPs after the detection of NDMA. Interestingly, Indian manufacturer Alembic Limited voluntarily withdrew its CEP in September 2018, following the nitroso impurity episode.
Table 3: List of Valsartan CEPs as of Sept. 17, 2018
U.S. Perspective
The FDA has different assessment criteria and methodology than the EU/EDQM. The guidance Changes to an Approved NDA or ANDA12 applies to changes made throughout the drug substance manufacturing process, i.e., from the starting material through the final drug substance.
Changes in key starting material manufacturing processes are left to the evaluation of the master file holder and, based on the evaluation, reporting criteria, and related documentation, shall be submitted in master files.
In section VII.B.5, the guidance indicates that “Changes in the synthesis or manufacture of the drug substance that may affect its impurity profile and/or the physical chemical or biological properties.”
The assessment of such changes should be done based on sections IV.A.1 and IV.A.2:
1. Conformance to Specifications
“Conformance to a specification means that the material, when tested according to the analytical procedures listed in the specification, will meet the listed acceptance criteria.”
2. Additional Testing
Evaluation of changes in the impurity or degradant profile could first involve profiling using appropriate chromatographic techniques and then, depending on the observed changes in the impurity profile, toxicology tests to qualify a new impurity or degradant or to qualify an impurity that is above a previously qualified level.
This guideline is enough to alert manufacturers to evaluate and assess the new impurities that may form during the changed process and their subsequent impact on the marketed substance and product.
In the new draft guidance Postapproval Changes to Drug Substances13 Section VIII.B (Changes in Route of Synthesis of One or More Steps) states:
In general, changes in route of synthesis are considered to have a moderate to high potential to adversely affect the impurity profile of the drug substance. The manufacturing process should be validated using the new route of synthesis. Impurity carryover studies and spike/purge studies should be conducted as appropriate. Control of mutagenic impurities in or expected to be in the final drug substance should be evaluated according to ICH M7 (section 4.1).
This new guideline explicitly mentions impact assessment as per ICH M7 for genotoxic impurities.
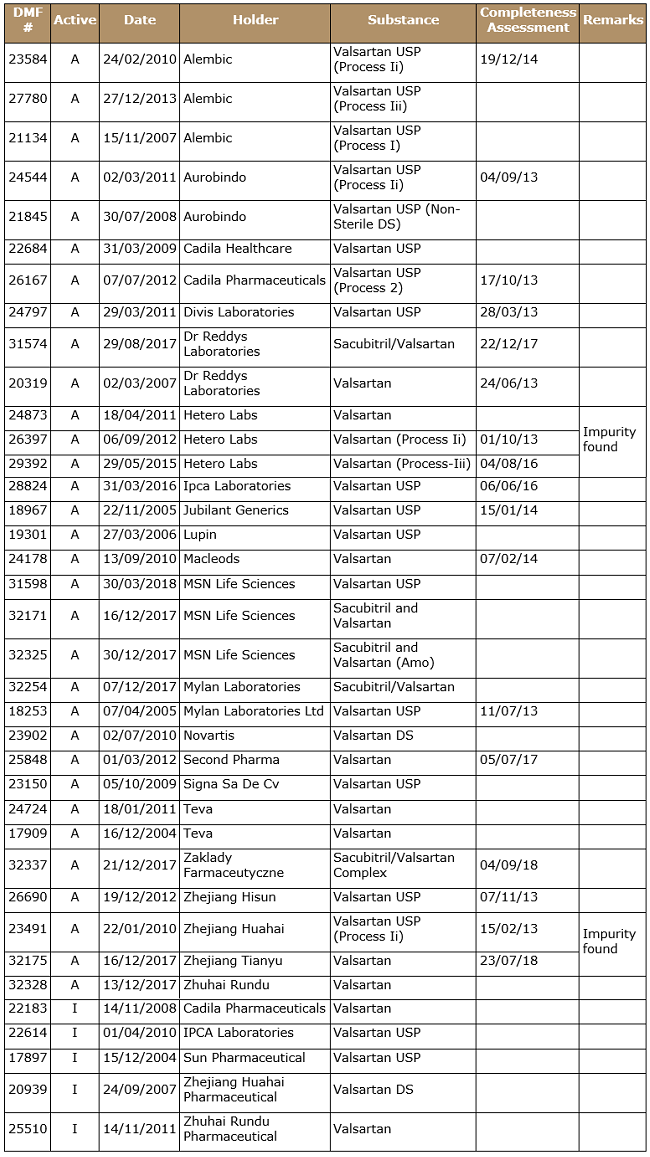
Table 4: List of USDMF as on Sep. 17, 2018
Assessment
Nitroso impurities are very well studied and documented in food industry and drinking water specifications worldwide. However, their origin and impact were not studied or extrapolated in pharmaceutical substance manufacturing processes.
I have reason to believe that all the submission holders have been updating their regulatory submissions according to the effective guidelines and that regulators have acted according to the prevalent knowledge database at their end.
As a recent article rightly pointed out,14 pharmaceutical substances can’t be and probably will not be analyzed for all the probable carcinogens on a regular basis because of limitations with pharmacopoeias, regulatory authorities, and the industry.
However, this unfortunate incident, which is still being extended to other ARBs (losartan, and irbesartan), forces us to look beyond the mere guidelines and processes to the experience of other industries, in this case nitroso impurities in the food and water industries.
This case may benefit some manufacturers that claim their products are “nitroso impurity free,” as well as analytical laboratories that have validated methods for analyses of nitroso impurities in drug substances.
To avoid cases similar to this in the future, the pharmaceutical industry may have to consider adopting a collaborative approach during the research, development, and manufacture of drug substances to absorb the knowledge available in other domains.
Notes:
- Valsartan is an angiotensin II receptor blocker (ARB) that treats high blood pressure and heart failure.
- From Toxnet: https://toxnet.nlm.nih.gov/
- The calculated acceptable intake for NDMA is based on methods described in the ICH Guidance M7(R1) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk (https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM347725.pdf)
- http://en.huahaipharm.com/news_det.asp?info_kind=002001&ID=698
- http://en.huahaipharm.com/news_det.asp?info_kind=002001&ID=699
- https://www.linkedin.com/wukong-web/articleShare/https%3A%2F%2Fwww.linkedin.com%2Fpulse%2Fhow-did-n-nitroso-dimethylamine-ndma-mm048709-get-moritz-perscheid?from=singlemessage&trk=article_share_wechat&ts=1532574566046
- https://www.newswire.com/news/update-on-valsartan-api-a-statement-from-the-company-20610371?from=singlemessage
- PA/PH/CEP (04) 2, 4R, December 2009
- Guideline On Requirements For Revision/Renewal Of Certificates Of Suitability To The European Pharmacopoeia Monographs, Pa/Ph/Cep (04) 2, 6r
- https://www.edqm.eu/sites/default/files/guideline-requirements-revision-renewal-cep-to-ph-eur-monographs-september2018.pdf
- http://en.huahaipharm.com/news_det.asp?info_kind=002001&ID=698
- https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm077097.pdf
- https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM619947.pdf
- http://www.pharmtech.com/after-valsartan-recalls-regulators-grapple-nitrosamine-contamination-apis
About The Author:
 Ketan Agravat is executive VP of RA and quality at OMSAV Pharma Research. He has more than 25 years of experience in the pharmaceutical industry, the last 15 in the field of regulatory intelligence and compliance for active pharmaceutical ingredients (APIs) and finished dosage forms (FDFs). Prior to joining OMSAV, Agravat served as VP of regulatory affairs and as a core team member for regulatory compliance and data integrity investigations at Unimark Remedies Limited. He has also held executive-level positions at Jubilant Life Sciences and Ind-Swift Laboratories Limited. Agravat has faced audits from FDA, EDQM (Europe), ANSM (France), ANVISA (Brazil), and TGA (Australia), and other regulators. He received his B.S. (science) and M.S. (organic chemistry) from Ravishankar University. You can reach him at ketanagravat@gmail.com or connect with him on LinkedIn.
Ketan Agravat is executive VP of RA and quality at OMSAV Pharma Research. He has more than 25 years of experience in the pharmaceutical industry, the last 15 in the field of regulatory intelligence and compliance for active pharmaceutical ingredients (APIs) and finished dosage forms (FDFs). Prior to joining OMSAV, Agravat served as VP of regulatory affairs and as a core team member for regulatory compliance and data integrity investigations at Unimark Remedies Limited. He has also held executive-level positions at Jubilant Life Sciences and Ind-Swift Laboratories Limited. Agravat has faced audits from FDA, EDQM (Europe), ANSM (France), ANVISA (Brazil), and TGA (Australia), and other regulators. He received his B.S. (science) and M.S. (organic chemistry) from Ravishankar University. You can reach him at ketanagravat@gmail.com or connect with him on LinkedIn.