
Pharmacy Compounding Vs. Manufacturing For Clinical Studies: The Regulatory Perspective
By Stephen Langille, senior microbiology consultant, ValSource, Inc.

Pharmacy compounding practices are regularly used by hospital and clinical center pharmacies for the preparation of parenteral drug products (i.e., compounded sterile products) for patient care. Many of these clinical pharmacies also manufacture drug products for use in clinical studies executed according to the methodology established in investigational new drug (IND) applications submitted to the FDA. However, FDA requirements and compendial standards for pharmacy compounding and outsourcing continue to evolve and the FDA has different requirements for products prepared through pharmacy compounding practices as compared to those manufactured for use in clinical studies. Understanding the nuances of pharmacy compounding and clinical manufacturing obligations is critical for ensuring regulatory compliance, product quality, and patient safety.
Pharmacy Compounding
Pharmacy compounding in the United States is subject to the regulations established in sections 503A and 503B of the Food Drug and Cosmetic (FD&C) Act. Drug products compounded in 503A facilities are, among other things, exempt from FD&C Act sections relating to current good manufacturing practices (section 501(a)(2)(B)), labeling requirements for adequate directions for use (section 502(f)(1)), and approval under new drug applications or abbreviated new drug applications (section 505).1 503A compounding also requires a prescription for the preparation of each compounded product for an identified individual and 503A facilities generally do not undergo FDA inspection unless there is a complaint.2, 3 Drug products compounded at 503B or “outsourcing” facilities are also exempt from FD&C Act sections 502(f)(1) and 505, as well as FD&C Act section 582 concerning drug supply chain track and trace requirements. However, 503B facilities do not require prescriptions to manufacture drug product. Additionally, they are required to register with the FDA as an outsourcing facility, comply with CGMPs as per FD&C Act section 501(a)(2)(B), and are subject to FDA inspections on a risk-based schedule.4, 5
Although the CGMP requirements differ for 503A and 503B facilities, drug product prepared at either type of facility will be deemed adulterated “if it has been prepared, packed, or held under insanitary conditions” as per section 501(a)(2)(A) of the FD&C Act. The FDA’s September 2018 draft guidance for industry titled Insanitary Conditions at Compounding Facilities provides an interpretation and examples of what might be considered insanitary conditions at these facilities.3 Several of the examples provided in the guidance differ from compendial recommendations and common pharmacy practice. Examples include the use of non-sterile gown components, the use of primary engineering controls or practices that do not prevent operators from contacting first air, the presence of sinks, drains, or water sources in an ISO 5 buffer room, and the use of non-sterile disinfecting agents in ISO classified areas. It is the expectation of FDA inspectors that insanitary conditions be avoided in order to comply with the FD&C Act even if compendial standards might advise otherwise.
Another difference between 503A and 503B sterile drug products is the mechanisms by which beyond use dates (BUDs) and in-use times are established. BUDs, as referenced in United States Pharmacopoeia (USP) chapter <797> Pharmaceutical Compounding – Sterile Preparations,6, 7 refer to the amount of time that a compounded sterile product can be stored based upon the microbiological, chemical, and physical risks to the product. BUDs for products manufactured at 503A compounding pharmacies are usually based upon the recommendations provided in USP chapter <797> related to the risks associated with the product, the compounding operation, and storage conditions. Although 503B facilities are subject to CGMPs, the expiration dating and stability test requirements of 21 CFR 211.166 and 21 CFR 211.137 may be waived if the BUD serves as the expiration date, the BUD is based on literature or scientific information, and the BUD is assigned based on the processing condition. Unpreserved product of aggregate batch sizes of ≤ 1,000 units manufactured by aseptic processing will generally not be subject to regulatory action if the BUD is no more than six days when stored at controlled room temperature or nine days if refrigerated. These storage limits are extended to 14 and 28 days, respectively, if the product is terminally sterilized, and the BUD for both terminally sterilized and aseptically processed products can be extended further if a passing sterility test is obtained prior to release. In addition, 503B compounded products requiring additional manipulation (e.g., dilution or reconstitution) prior to use should include the maximum post-manipulation in-use time in order to fulfill the requirement of FD&C Act section 503B(a)(10)(B). However, data to support the conditions of in-use hold times would only be necessary if they meet or exceed 4 hours at room temperature or 24 hours if refrigerated.8
BUDs are usually not applied to sterile products manufactured for use in clinical studies. This is primarily because the results of sterility testing are normally required prior to the release of the investigational drug for use in clinical studies and the product shelf life is addressed in the stability protocol submitted in the IND application.9, 10 The amount of data required to support product stability is commensurate with the phase of clinical study and a shelf life well in excess of typical BUDs is the norm when supporting data is provided.10, 11 However, exceptions for sterile clinical study product BUDs without the submission of extensive supporting data may be possible for bespoke product for use in small clinical studies if adequate control of the starting materials and compounding process is established and the BUD is based upon scientific principles and/or literature. The manufacturing processes, product specifications, and any diluted or reconstituted product in-use times for all clinical study material should be clearly described in the regulatory submission.
Clinical Study Manufacturing Processes
Drug products for use in clinical studies must comply with CGMPs and the manufacturing processes established in the IND application. However, requirements for the extent of manufacturing control and adherence to CGMPs will depend upon the phase of clinical studies. For instance, Phase 1 clinical study material is subject to some, but not all, of the CGMPs established in 21 CFR 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals. 21 CFR 210.2(c) states that “An investigational drug for use in a Phase 1 study…is exempt from compliance with the regulations in part 211 of this chapter. However, this exemption does not apply to an investigational drug for use in a Phase 1 study once the investigational drug has been made available for use by or for the sponsor in a Phase 2 or Phase 3 study…or the drug has been lawfully marketed.” In other words, if the Phase 1 drug has graduated to inclusion in Phase 2 or 3 studies or been marketed, it would then be subject to all CGMP regulations provided in 21 CFR 211. The FDA’s 2008 guidance for industry titled CGMP for Phase 1 Investigational Drugs provides recommendations that manufacturers of Phase 1 investigational drugs can use to comply with the statutory requirement for CGMP under section 501(a)(2)(B) of the FD&C Act.5 The information to be provided in Phase 1 clinical study applications, including appropriate finished product tests (such as sterility and endotoxins tests), is provided in the 1995 guidance for industry titled Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-derived Products.10
The exemption provided in 21 CFR 210.2(c) for investigational drugs used in Phase 1 studies does not extend to Phase 2 and 3 clinical study drugs. Manufacturing consistency and reproducibility are critical to demonstrate clinical effectiveness in a larger patient population and it is difficult, if not impossible, to evaluate drug product safety and efficacy if there is batch to batch variation in the manufacturing process. Thus, the implementation and execution of CGMPs become increasingly important as the phases of clinical studies progress and additional patients and product are included in the studies. Likewise, the level of FDA scrutiny regarding adherence to the CGMPs as stated in 21 CFR 211 will increase as clinical studies progress, and the FDA has published additional guidance documents detailing the CGMP and submission requirements for Phase 2 and 3 clinical studies.11, 12 Although FDA inspections for clinical study materials are infrequent, phase-appropriate compliance with CGMPs is expected. The table below summarizes some of the requirements and guidance documents related to clinical study drug manufacturing and compounding operations in the United States.
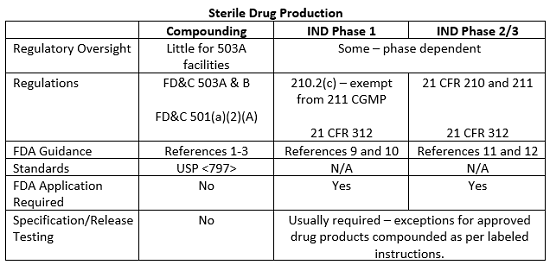
This article summarizes some of the major the differences between sterile compounding in 503A and B facilities and manufacturing practices for drug products for use in clinical studies. The line between compounding and manufacturing is sometimes blurred, and compendial and regulatory guidance on the subject, though steadily evolving, is still in a state of flux. However, continued collaboration between clinical, pharmacy, and regulatory stakeholders should allow for the finalization of standards and guidance documents designed to ensure product quality and patient safety without limiting the clinical flexibility that pharmacy compounding practices afford or affecting the quality and availability of products used in clinical studies.
References:
- Draft Guidance for Industry: Hospital and Health System Compounding Under the Federal Food, Drug, and Cosmetic Act. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Office of Compliance/OUDLC. April 2016.
- FDA Guidance for Industry: Pharmacy Compounding of Human Drug Products Under Section 503A of the Federal Food, Drug, and Cosmetic Act. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). June 2016.
- FDA Draft Guidance for Industry: Insanitary Conditions at Compounding Facilities. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research Office of Compliance. September 2018.
- FDA Guidance for Industry: Facility Definition Under Section 503B of the Federal Food, Drug, and Cosmetic Act. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research. May 2018.
- FDA Guidance for Industry: For Entities Considering Whether to Register as Outsourcing Facilities Under Section 503B of the Federal Food, Drug, and Cosmetic Act. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). August 2015.
- USP Chapter <797> Pharmaceutical Compounding – Sterile Preparations. Official May 1, 2018. USP 42-NF37.
- USP <797> Pharmaceutical Compounding – Sterile Preparations. Official implementation date of December 1, 2019 postponed pending appeals.
- Draft Guidance for Industry: Current Good Manufacturing Practice—Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research. December 2018.
- FDA Guidance for Industry: CGMP for Phase 1 Investigational Drugs. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory Affairs (ORA). July 2008.
- Guidance for Industry: Content and Format of Investigational New Drug Applications for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-derived Products. Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER). November 1995.
- Guidance for Industry: INDs for Phase 2 and Phase 3 Studies: Chemistry Manufacturing and Controls Information. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). May 2003.
- Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory Affairs (ORA). September 2004.
About The Author:
 Stephen Langille received his B.S. in biology from the University of Massachusetts and his Ph.D. in microbiology from the University of Maryland. He worked for the FDA for 19 years as a microbiology reviewer, branch chief, and director of the Center for Drug Evaluation and Research’s Division of Microbiology Assessment. He has served on several USP expert committees to develop and improve compendial standards for microbiological and particulate matter quality in injectable products. He currently works as a senior microbiology consultant at ValSource, Inc.
Stephen Langille received his B.S. in biology from the University of Massachusetts and his Ph.D. in microbiology from the University of Maryland. He worked for the FDA for 19 years as a microbiology reviewer, branch chief, and director of the Center for Drug Evaluation and Research’s Division of Microbiology Assessment. He has served on several USP expert committees to develop and improve compendial standards for microbiological and particulate matter quality in injectable products. He currently works as a senior microbiology consultant at ValSource, Inc.