
Pinpointing Supply Chain Risk Through Line-Of-Sight Shipping Validation
By Chris M. Hanff, CMQ/OE, ValSource, LLC

Shipping and its validation are too often underappreciated in pharmaceutical quality systems (PQS), which can hide risk and uncertainty in the supply chain. A line-of-sight (LOS) approach to shipping validation brings focus to risks in transportation, and rouses quality improvement opportunities throughout the PQS. An LOS approach begins with the end in mind — connecting product critical quality attributes (CQAs) to integrity of the cold chain and to shipping validation design.
When is your product more at risk than when it’s out of your hands and out of your control? What will you do about it?
The purpose of this article is to express the importance and value of the shipping process and its validation. Shipping validation can even act as a vehicle to deliver improvement through the entire pharmaceutical quality system (PQS). This article is aimed at the product-patient dynamic, discussing points where shipping validation intersects the PQS.
This article is a product of my work in and around shipping and validation involving passive and active transport of pharmaceutical finished goods, raw materials, and single-use disposable assemblies, during which I’ve engaged with third-party laboratories, logistics providers, and general supply chain partners that had external partnerships with pharmaceutical drug product license holders. Through these experiences, a common theme — and maybe a truism — emerged about shipping validation: Shipping and its validation are out of sight or overlooked, which hides risk and uncertainty in the supply chain.
Is Shipping And Its Validation Out Of Sight (And Out Of Mind)?
Shipping validation is a deliverable with new product commercialization, and we in pharma seem to recognize that. But does shipping validation get the requisite focus? Compared with validation of the drug product manufacturing process, qualification of materials and vendors, and product and process scale-up and transfer, validation of the shipping process has hardly a voice. Instead, shipping validation is often overlooked or is an afterthought.
Some pharma companies are even strategizing to defer shipping validation work as a post-market commitment (PMC). One might argue that routine, commercial drug product transportation has acceptable risk. Or, they might argue that shipping is a simple endeavor, and that there is much confidence or certainty in shipping. This rings true in our daily lives, doesn’t it? We shop online, and speedy shipment in one to two days is the norm. We don’t give a second thought to shipping reliability. So, I believe we project those consumer experiences onto the commercial drug product shipping process. I suggest we do so blindly, based on my experience that pharma doesn’t scrutinize its shipping and distribution enough until something goes wrong.
Why is it that one of the most accessible places to see pharma products — the warehouse and dock — is also one of the least visited by staff? Pharma quality auditors typically visit the warehouse dock when they inspect vendor or partner warehouse space. How many of us get down to our own docks? If we don’t, aren’t we then ignorant of the shipping process, at least more so than the product manufacture process? Indeed, the technical challenges about shipping are simpler than drug manufacture. Yet if we don’t invest in solving even simple problems, potentially significant effects could catch us unprepared.
A Line-Of-Sight Approach
Shipping validation, though generally simpler than manufacture process validation, still warrants an effective and efficient approach to assure product quality and availability. A right-sized approach to pharma product shipping validation can deliver both effectiveness and efficiency. A line‑of‑sight (LOS) approach aids right-sizing by keeping focus on what’s important to shipping validation. How? Let’s first review the conventional definition for LOS: a straight line along which an observer has unobstructed vision. That vision is unobstructed is an important point.
Think of LOS as looking through a scope (telescope, microscope, or periscope), and recognize how your field of view is exclusively on the object, or target, of your vision. Each scope offers line‑of‑sight by removing all potential distractions from your field of view. That’s dialing in LOS with the coarse focus, but let’s use the fine focus to get more precise. A line‑of‑sight approach means you can extend an imaginary straight line to the work in validation at Stage 1 (process design), Stage 2 (process qualification), and Stage 3 (continued process verification)?1
In the LOS approach, what is that ultimate focus? It’s a vision we all share in pharma: safe and effective products reliably delivered to patients. So, we can consider critical quality attributes (CQAs) plus delivery as the ultimate focus. Delivery of safe and effective product is where our work culminates, and it’s where we anchor our line‑of‑sight.
Beginning with the end in mind is integral to the LOS approach. The product-patient dynamic is the endpoint in our pharma work stream. That dynamic includes the patient; the product and its quality, availability, and delivery timing; and administering product to patient. Anchoring LOS at the product-patient dynamic focuses our scope on what’s ultimately important and leaves out of focus what’s not as important.
LOS And The Status Quo
In early drug development, pharma gains practice by shipping developmental product to off‑site clinical institutions, and often to off-site laboratories. In early developmental product shipments, it’s easy to monitor the transportation with high levels of attention and vigilance. In the same early developmental shipments, it’s easy to use familiar shipping solutions for cold-chain transport (e.g., packages utilizing insulated sides, cold charge [gel packs, phase change materials, dry ice, etc.]). It’s also easy to delay any decisions affecting the transportation of commercial drug product until commercialization — and to just drop in a time and temperature data logger in the meantime. The common argument against early adoption of shipping validation decisions is that too many final product parameters are not yet known. This is questionable rationale.
By the time clinical trials have commenced, I would suggest that enough product attributes are well-known to design a user requirements specification (URS) for the shipping system(s). Consider some product attributes relevant to shipping that are needed to prepare a URS for a shipping system (Table 1).
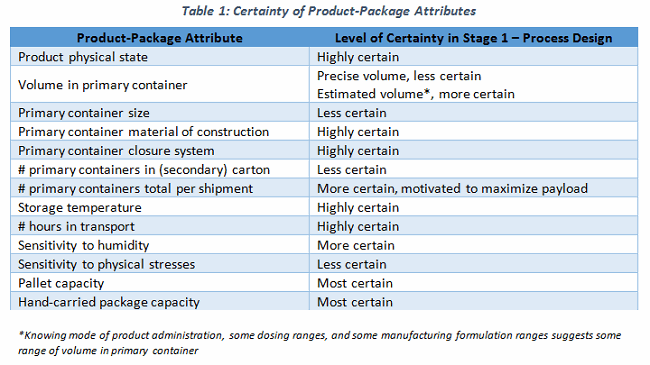
It would be simple to depict the design space for the shipping process, and to create a URS with which to identify and select the appropriate shipping system(s). Yet, you rarely see a URS supporting the use of a selected cold chain shipper, or for an active control pallet container. Sure, there are variables (like volume in container) and a cascade of additional dependent variables (Table 2). These can be determined, if not approximated, reasonably easily.

It could be daunting to devise a shipping URS or a shipping strategy with so many variables undetermined during product development. Instead of working from the many unknowns (variables) and extrapolating or devising a future shipping system, work with the end in mind! There are some established shipping constraints, such as pallet dimensions, that actually help to define our variables. To illustrate the concept, consider Russian nesting dolls, where a painted wooden doll is opened at its waist to reveal inside another, smaller doll. This doll-within-a-doll model is analogous to our shipping system. If we assume there is a physical size constraint for the outermost doll, the size and number of dolls within can be determined with a high degree of certainty.
We can use the dimensional constraint for a standard single pallet (or, alternately, a Euro pallet). Only so many boxes of a given size can fit within the standard width, depth, and height constraints of a standard pallet. There’s a science to this called “pallet cubing” or “cubing out.” Figuring out the given box size is important but is at best secondary to understanding what proportion of the pallet space will be for payload, for dunnage, and for temperature control materials such as insulation and cold charge.
Again, the LOS approach works with the end in mind. To ensure product temperature is maintained throughout, we first must determine how much of the pallet space should be spent on the cold charge. This comes first because the cold charge quantity is dependent on time and differential ambient temperature, and is largely independent of payload and dunnage. It’s noteworthy that once contracted, a third-party logistics (3PL) provider will share their proprietary equations regarding how much cold charge is needed (seasonally) for shipments as a function of the shipment duration, ambient temperature, time out of refrigeration, and timing/ opportunity to replenish the cold charge (dry ice pellets/ blocks, gel packs, phase change materials, etc.). So, subtract from the pallet space how much cold charge is required to maintain temperature for the trip. The remaining pallet space is available to payload and dunnage.
Dunnage can be highly customized, limiting generalizations about the quantity needed to protect primary packaging. A recommended practice is using excess dunnage until simulated shipping physical stress tests demonstrate how much dunnage is needed to safeguard the physical integrity of the primary package and product within. Following this LOS approach, it’s possible to cube out the pallet and maximize the payload, after first allocating enough space to the cold charge and then to dunnage. This systematic process for pallet shipments applies to single package systems, as well. In both scenarios, a URS could be drafted despite unknown commercial product-package attributes.
LOS At Third Parties
Drug product shipping and distribution are routinely outsourced to one of a handful of cold chain couriers, and almost blind-faith acceptance is placed in the product distribution network, or supply chain — whether the 3PL is known or unknown. It is common to find service level agreements (SLAs) between pharma companies and their 3PLs. Most SLAs define contractual obligations about volume of goods moved, timing of movement, and economized rates for the movement. Yet, it is uncommon for quality agreements to be in place in these same contracted relationships; even more scarce are on‑site quality audits of 3PLs. Again I ask, “Why not?” Pharma puts its products in the custody of 3PL partners to provide safe carriage and holding of its products, most of which are quite sensitive to temperature and physical stresses.
From a compliance perspective, FDA “regards extramural facilities as an extension of the manufacturer’s own facility”2 and “recommends that owners and contract facilities implement written quality agreements as tools to delineate manufacturing activities for ensuring compliance with CGMP.”3 Additionally, ICH Q7 recommends a “written and approved contract” between manufacturers and their contractors that “defines in detail the GMP responsibilities, including the quality measures, of each party.”4 Beyond compliance requirements, it’s certainly simpler to find common ground on a proactively prepared quality agreement than it is to wrangle over quality issues once they are at hand, or even retrospectively. Through line-of-sight shipping validation, pharma companies can uncover previously unknown couriers and/or waypoints involved in drug product transportation, conditions that could be precluded or disclosed if a quality agreement is in place.
So again, I ask you: When is your product more at risk than when it’s out of your hands (control)? And what will you do about it?
About the Author:
 Chris M. Hanff, CMQ/OE, is a senior consultant in the pharma industry who teaches, writes, and provides his expertise and insights as an architect of effective quality and business systems. Hanff promotes his vision of “a pharma industry investing intentionally and proactively in quality” through a strategy of “teaching tools and techniques enabling knowledge transfer and cost of quality for deliberate, disciplined, decision making.” Hanff has nearly 20 years’ experience in biopharmaceutical operations, quality systems, and supply chain management building right-sized and reliable operating processes with line-of-sight focus on the product-patient dynamic across the entire value chain.
Chris M. Hanff, CMQ/OE, is a senior consultant in the pharma industry who teaches, writes, and provides his expertise and insights as an architect of effective quality and business systems. Hanff promotes his vision of “a pharma industry investing intentionally and proactively in quality” through a strategy of “teaching tools and techniques enabling knowledge transfer and cost of quality for deliberate, disciplined, decision making.” Hanff has nearly 20 years’ experience in biopharmaceutical operations, quality systems, and supply chain management building right-sized and reliable operating processes with line-of-sight focus on the product-patient dynamic across the entire value chain.
Connect with Chris at:
www.LinkedIn.com/in/chrishanff
- FDA Guidance for Industry, Process Validation: General Principles and Practices, 2011
- 21CFR 200.10
- FDA Guidance for Industry, Contract Manufacturing Arrangements for Drugs: Quality Agreements, 2016
- ICH Q7, Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients, 2016