
Protein Degraders: Progress, Limitations, & Future Directions
By Weijun Gui and Thomas Kodadek, The Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology

There is great excitement surrounding the development of small molecules that trigger the degradation of proteins. Intense activity in this area has resulted in the development of degraders targeting many different proteins and a few compounds are entering clinical trials. However, the limitations of current technology are increasingly apparent. This article will highlight these limitations and discuss potential emerging solutions.
In 2001, Crews, Deshaies, and colleagues reported the concept and initial development of PROTACs (proteolysis targeting chimeras).1 PROTACs are chemical dimerizers that force an E3 ubiquitin (Ub) ligase and a target protein (TP) into close proximity. Under favorable circumstances (see below), this results in poly-ubiquitylation of the TP and its subsequent destruction by the 26S proteasome (Fig. 1).2
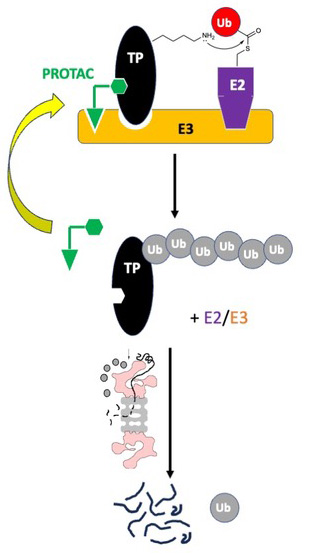
Figure 1: Schematic of the mechanism of action of a PROTAC.
The PROTAC concept was revolutionary in the sense that it envisioned that a small molecule could act as a catalytic inhibitor.3 In other words, by triggering a post-translational modification of the TP, a single PROTAC molecule could mediate the inactivation of multiple TP molecules. This has been described as “event-driven” inhibition via a hit and run mechanism. Presumably, this will lower the bar for the chemical matter required to engage the TP relative to the requirements for a conventional “occupancy-driven” inhibitor, since the formation of a tight, long-lived small molecule-TP complex is no longer required for TP inactivation. This raised hopes of drugging proteins that lack the deep pockets typically required for high affinity engagement by a small molecule. Another potentially enormous advantage of PROTACs is that the TP ligand need not be an inhibitor and thus can bind a surface of the TP far from the active site. This has huge potential ramifications for simplifying the development of highly selective degraders of specific members of multi-protein families in which the active sites of many family members are highly homologous and thus challenging to target selectively with traditional occupancy-driven antagonists.
Despite the great promise of PROTAC technology, the idea did not significantly impact drug development for several years. This was due to the lack of drug-like ligands for E3 ubiquitin ligases (the first PROTACs contained peptidic ligands representing native degrons). This changed with the development of a synthetic cell permeable peptidomimetic ligand for the von Hippel Landau (VHL) E3 Ub ligase (Fig. 2A), which enabled the creation of cell permeable PROTACs.4,5 Another seminal advance was the discovery that the closely related small molecules thalidomide and lenalidomide (Fig. 2A) are ligands for the E3 Ub ligase Cereblon (CRBN) that “reprogram” it to recognize certain “neo-substrates” (i.e., proteins that are not normally operated upon by CRBN), triggering their proteasome-mediated destruction.6-8 Since the discovery of these cell permeable E3 Ub ligase ligands, there has been an explosion of activity in the creation of PROTACs containing these molecules that target many different TPs, culminating with the recent development of the first clinical candidates by the biotechnology company Arvinas (Fig. 2B).
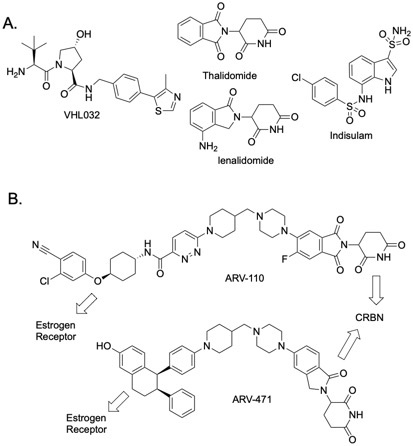
Figure 2: Structures of important molecules in degrader technology.
(A.) VHL ligand (VH032), two CRBN ligands (thalidomide and lenalidomide), and a MGD (indisulam).
(B.) Structures of the first two PROTACs to enter clinical trials.
Drawbacks Of Bivalent Inhibitors
While PROTACs are potentially revolutionary, their unique mechanism of action introduces some challenges. One issue is that PROTAC-driven TP-E3 Ub ligase proximity is necessary but not sufficient for efficient TP degradation. Studies of the proteome-wide effect of PROTACs containing promiscuous kinase ligands showed clearly that the degree of PROTAC-mediated degradation of a particular kinase could not be predicted by its affinity for the PROTAC.9,10 Many kinases engaged with high affinity by the PROTAC were not degraded and some that bound the PROTAC with only modest affinity were degraded efficiently. This was explained by subsequent structural and biophysical studies that revealed potent PROTACs must drive the formation of ternary complexes in which there are significant “neo-protein-protein interactions” (neo-PPIs) between the E3 and the TP (Fig. 1).11 Moreover, there must be a good alignment of the acceptor lysine on the TP and the activated Ub ester in the E2/E3 complex (Fig. 1). Otherwise, TP ubiquitylation will not occur. For this reason, extensive linker optimization is generally required to produce a potent PROTAC, which increases the difficulty and the expense of their development.
Another complication is that bivalent molecules such as PROTACs exhibit a nontraditional dose-response curve in which their activity reaches a peak at a certain concentration but then decreases at higher concentrations. This so-called hook effect is the result of high concentrations of the PROTAC driving the formation of dimeric PROTAC-E3 Ub ligase and PROTAC-TP complexes, which compete with formation of the all-important ternary complexes necessary to trigger TP poly-ubiquitylation.12 The hook effect may complicate the use of PROTACs pharmacologically.
A third issue is that most PROTACs are large molecules with non-optimal drug-like properties, including modest cell permeability. For example, a PROTAC comprised of JQ1, an inhibitor of the BET proteins, linked to the VHL peptidomimetic ligand, was found to be 165,000-fold less cell permeable than JQ1 itself.13 While this is likely an extreme example, and the potency of these catalytic PROTACs partially makes up for low permeability, there is great interest in improving the pharmacological properties of PROTACs.
Finally, for oncology and anti-infective applications, it is likely that the spectrum of mutations that create resistance to PROTACs will be quite different than that of a traditional inhibitor. The good news is that PROTACs should be less vulnerable to mutations in the TP that weaken the affinity of the PROTAC for the TP.14,15 This is because of the aforementioned fact that extremely high affinity and the formation of long-lived inhibitor-TP complexes are not essential for PROTAC function. The bad news is that mutations in the components of the E3 Ub ligase complex will lead to PROTAC insensitivity.16,17 Since very few E3 Ub ligases are essential proteins this is likely to be a significant problem clinically.
Emerging Strategies To Address The Limitations Of PROTACs
The above section should not be interpreted as a suggestion that PROTACs are deeply flawed. Far from it. Current PROTAC technology is good enough that, with sufficient resources and effort, compounds with acceptable activity and pharmacokinetic properties can certainly be developed and many PROTACs are likely to enter the clinic over the next few years. For example, optimization of the geometry and physicochemical properties of the linker in a PROTAC can have a profound effect on its activity and pharmacology.18,19 Identifying optimal linkers more rapidly and efficiently is likely to become easier over the next few years. Molecular modeling, perhaps driven by advances in computational/AI/machine learning methodology, will likely have a major impact. Another promising avenue is the development of combinatorial libraries of relatively stiff linkers to which TP and E3 Ub ligase ligands can be attached. These libraries can then be tested in cell-based assays to identify the most potent degraders.
Nonetheless, many investigators are exploring the development of radically different kinds of degraders in an effort to surmount the limitations of traditional PROTACs. A major focus is the search for “molecular glue”-type degraders (MGDs). MGDs, like PROTACs, are also chemical dimerizers that trigger TP degradation by localizing it to an E3 Ub ligase, but they are monovalent, more drug-like compounds. As such they tend to be more cell permeable and do not suffer the complications of the hook effect. The first such glues were thalidomide and its analogues (Fig. 2). As mentioned above, they bind to the E3 Ub ligase Cereblon and drive a conformational change that causes it to recognize neo-substrates. Another example is indisulam (Fig. 2), a low molecular weight, indole-containing sulfonamide. It glues together the E3 ligase DCAF15 and the splicing factor RBM39, causing poly-ubiquitylation and destruction of the latter.20
While MGDs are highly sought after, they are hard to find. All existing MGs were discovered in phenotypic screens, and it was only realized much later that they function as degraders. Several companies are now focused on establishing general and reliable methods to discover MGDs,21 including Plexium, Monte Rosa, Triana, Degron, Evotec, Proxygen, and others. One approach is to establish high-throughput screens to identify molecules capable of bringing an E3 Ub ligase and a TP into close proximity. However, while biochemical assays to do so exist and can accommodate very large libraries, it is unclear what percentage of compounds that assemble with the TP and an E3 Ub ligase into a ternary complex formation will prove to be good degraders. As mentioned above, proximity is necessary, but not sufficient, for TP degradation. Another potential issue is that screens for ternary complexes will most easily find compounds that support the formation of quite stable assemblies, yet this may not be desirable for efficient TP degradation, since much of the potency of degraders comes from their catalytic mechanism of action. It is easy to imagine that turnover would be compromised by an overly stable ternary complex. Finally, a major issue in developing biochemical assays for MGD discovery is to decide which E3 Ub ligase to use. There are hundreds in the human proteome and it is very difficult to predict, a priori, which of these will “play well” with a particular TP.
Given these issues, there is also great interest in setting up high-throughput, cell-based screens for MGDs, for example by monitoring small molecule-triggered degradation of a TP-GFP fusion protein.22 The advantage of this approach is that it theoretically brings into play all of the E3 ligases expressed in the cell. It also has the advantage of screening for exactly what one wants (degradation), rather than something close to what one wants (ternary complex formation). This approach may be most effective using custom screening collections in which each molecule has a “toehold” on the TP, for example by incorporating into the library synthesis an invariant, low molecular weight TP-binding fragment.
A more radical variant of PROTAC technology was introduced recently by Barany, Kodadek, and co-workers.23 They described the creation of “SAPTACs” (self-assembled proteolysis-targeted chimeras). A SAPTAC is a bivalent degrader in which the E3 Ub ligase and TP ligands are connected by a rapidly reversible linkage, such as, for example, a boronic ester produced by the reversible condensation of a 1,2-diol and a boronic acid (Fig. 3A). The idea behind SAPTACs is that the smaller component pieces could more easily diffuse through the membrane than a stably linked PROTAC, then reversibly assemble inside the cell with the linked equilibria being drawn into the ternary complex thanks to the stabilizing effects of the neo-PPIs (Fig. 3A).
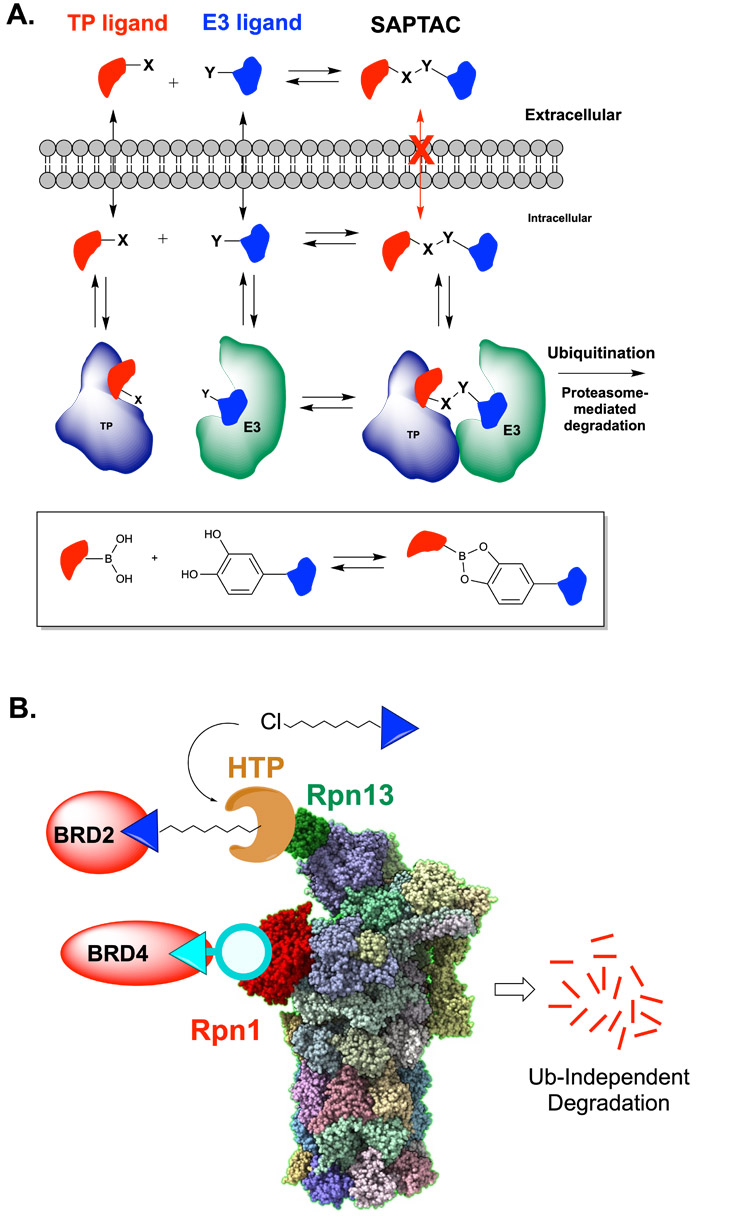
Figure 3: Alternative PROTAC designs.
(A.) SAPTACs (self-assembled PROTACs) contain functional groups capable of rapidly reversible covalent coupling, such as the formation of a boronic ester from a boronic acid and catechol (see box).
(B.) Ubiquitin-independent TP degradation through direct to proteasome recruitment. Degradation of native BET proteins has been achieved by either recruitment of Brd2 to an engineered Rapn13-HaloTag protein (HTP) fusion protein by a ligand-chloroalkane conjugate and through recruitment of Brd4 to Rpn1 via ligand-macrocyclic peptide conjugate.
The SAPTACs reported were slightly less potent than analogous irreversibly linked PROTACs due to the relatively poor permeability of the functional groups used to create the reversible linkage (especially the boronic acid). Nonetheless, future elaborations of this idea using more cell permeable functional groups allow this approach to realize its potential. A major advantage of SAPTACs is that they do not exhibit a hook effect since there is never a high concentration of the bivalent molecule present to compete with ternary complex formation.23
Another major divergence from PROTAC orthodoxy is the idea that degraders can be made that short-circuit the requirement for E3 ubiquitin ligases (Fig. 3B). Mechanistic studies have shown that the proteasome is capable of degrading non-ubiquitylated proteins so long as these substrates are trafficked to the proteasome in some other way.24-26 A group at Genentech recently published the first example of a ubiquitin-independent degrader (UID).27 They employed mRNA display to find a macrocyclic peptide ligand for the proteasome component Rpn1 (PSMD2). When a BET protein ligand was attached to this peptide, and the chimera was added to permeabilized cells, modest degradation of Brd4 was observed without the requirement for its ubiquitylation. In a similar vein, our group has recently shown that in cells engineered to express a fusion of HaloTag protein (HTP) to Rpn13 (one of the Ub receptors in the proteasome), chloroalkane-JQ1 conjugates can drive the turnover of Brd2 in a ubiquitin-independent fashion.28
In another approach that can eliminate the requirement for E3 Ub ligases, Nomura and colleagues used a cell-based screening approach to identify a covalent molecule that inhibits the proliferation of leukemia cells in a proteasome-dependent fashion. Remarkably, chemoproteomic analysis revealed that this small molecule engages an E2, not an E3, Ub ligase called UBE2D and drives the poly-ubiquitylation and subsequent degradation of NF-kB.29
Much more work will be necessary to evaluate the utility of UIDs and E2-dependent degraders. A potentially attractive feature of these degraders is that resistance obviously could not arise though elimination of E3 activity through mutation. It may be more difficult for cancer cells to generate resistance to UIDs and E2-targeted PROTACs because almost all of the proteasome sub-units and E2 Ub ligases are encoded by essential genes. However, very few ligands suitable for the construction of such PROTACs are currently available. Another concern is that it has been reported that poly-Ub chains stimulate the activity of the proteasome30,31 and it remains to be determined if this will limit the activity of UIDs (though for E2-targeted PROTACs this will not be an issue).
Conclusion
Targeted protein degradation is a drug development strategy that holds great promise. The first generation of small molecule degraders to enter the clinic, mostly in the oncology space, will be E3 Ub ligase targeted PROTACs. The discovery of novel MGDs will continue to be an area of heavy focus and investment and will likely result in the development of clinical candidates within the next five years. At the same time, SAPTACs, UIDs, E2-targeted degraders, and other novel PROTAC variants will be thoroughly vetted. It will be interesting to see if any of these approaches rise to clinical relevance.
References
- Sakamoto, K.M., Kim, K.B., Kumagai, A., Mercurio, F., Crews, C.M., and Deshaies, R.J. (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA 98, 8554-8559. 10.1073/pnas.141230798.
- Békés, M., Langley, D.R., and Crews, C.M. (2022). PROTAC targeted protein degraders: the past is prologue. Nature Reviews Drug Discovery 21, 181-200. 10.1038/s41573-021-00371-6.
- Bondeson, D.P., Mares, A., Smith, I.E., Ko, E., Campos, S., Miah, A.H., Mulholland, K.E., Routly, N., Buckley, D.L., Gustafson, J.L., et al. (2015). Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 11, 611-617. 10.1038/nchembio.1858.
- Buckley, D.L., Gustafson, J.L., Van Molle, I., Roth, A.G., Tae, H.S., Gareiss, P.C., Jorgensen, W.L., Ciulli, A., and Crews, C.M. (2012). Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew Chem Int Ed Engl 51, 11463-11467. 10.1002/anie.201206231.
- Buckley, D.L., Van Molle, I., Gareiss, P.C., Tae, H.S., Michel, J., Noblin, D.J., Jorgensen, W.L., Ciulli, A., and Crews, C.M. (2012). Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc 134, 4465-4468. 10.1021/ja209924v.
- Ito, T., Ando, H., Suzuki, T., Ogura, T., Hotta, K., Imamura, Y., Yamaguchi, Y., and Handa, H. (2010). Identification of a Primary Target of Thalidomide Teratogenicity. Science 327, 1345-1350. 10.1126/science.1177319.
- Lu, G., Middleton, R.E., Sun, H., Naniong, M., Ott, C.J., Mitsiades, C.S., Wong, K.-K., Bradner, J.E., and Kaelin, W.G. (2014). The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 343, 305-309. 10.1126/science.1244917.
- Krönke, J., Udeshi Namrata, D., Narla, A., Grauman, P., Hurst Slater, N., McConkey, M., Svinkina, T., Heckl, D., Comer, E., Li, X., et al. (2014). Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 343, 301-305. 10.1126/science.1244851.
- Bondeson, D.P., Smith, B.E., Burslem, G.M., Buhimschi, A.D., Hines, J., Jaime-Figueroa, S., Wang, J., Hamman, B.D., Ishchenko, A., and Crews, C.M. (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol 25, 78-87 e75. 10.1016/j.chembiol.2017.09.010.
- Huang, H.T., Dobrovolsky, D., Paulk, J., Yang, G., Weisberg, E.L., Doctor, Z.M., Buckley, D.L., Cho, J.H., Ko, E., Jang, J., et al. (2018). A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem Biol 25, 88-99 e86. 10.1016/j.chembiol.2017.10.005.
- Gadd, M.S., Testa, A., Lucas, X., Chan, K.-H., Chen, W., Lamont, D.J., Zengerle, M., and Ciulli, A. (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chemical Biology 13, 514-521. 10.1038/nchembio.2329.
- Douglass, E.F., Miller, C.J., Sparer, G., Shapiro, H., and Spiegel, D.A. (2013). A Comprehensive Mathematical Model for Three-Body Binding Equilibria. Journal of the American Chemical Society 135, 6092-6099. 10.1021/ja311795d.
- Foley, C.A., Potjewyd, F., Lamb, K.N., James, L.I., and Frye, S.V. (2020). Assessing the Cell Permeability of Bivalent Chemical Degraders Using the Chloroalkane Penetration Assay. ACS Chem Biol 15, 290-295. 10.1021/acschembio.9b00972.
- de Wispelaere, M., Du, G., Donovan, K.A., Zhang, T., Eleuteri, N.A., Yuan, J.C., Kalabathula, J., Nowak, R.P., Fischer, E.S., Gray, N.S., and Yang, P.L. (2019). Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nature Communications 10, 3468. 10.1038/s41467-019-11429-w.
- Burke, M.R., Smith, A.R., and Zheng, G. (2022). Overcoming Cancer Drug Resistance Utilizing PROTAC Technology. Frontiers in Cell and Developmental Biology 10. 10.3389/fcell.2022.872729.
- Zhang, L., Riley-Gillis, B., Vijay, P., and Shen, Y. (2019). Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Molecular Cancer Therapeutics 18, 1302-1311. 10.1158/1535-7163.MCT-18-1129.
- Hanzl, A., Casement, R., Imrichova, H., Hughes, S.J., Barone, E., Testa, A., Bauer, S., Wright, J., Brand, M., Ciulli, A., and Winter, G.E. (2022). Charting functional E3 ligase hotspots and resistance mechanisms to small-molecule degraders. bioRxiv, 2022.2004.2014.488316. 10.1101/2022.04.14.488316.
- Bashore, F.M., Foley, C.A., Ong, H.W., Rectenwald, J.M., Hanley, R.P., Norris-Drouin, J.L., Cholensky, S.H., Mills, C.A., Pearce, K.H., Herring, L.E., et al. (2023). PROTAC Linkerology Leads to an Optimized Bivalent Chemical Degrader of Polycomb Repressive Complex 2 (PRC2) Components. ACS Chemical Biology 18, 494-507. 10.1021/acschembio.2c00804.
- Poongavanam, V., Atilaw, Y., Siegel, S., Giese, A., Lehmann, L., Meibom, D., Erdelyi, M., and Kihlberg, J. (2022). Linker-Dependent Folding Rationalizes PROTAC Cell Permeability. Journal of Medicinal Chemistry 65, 13029-13040. 10.1021/acs.jmedchem.2c00877.
- Han, T., Goralski, M., Gaskill, N., Capota, E., Kim, J., Ting, T.C., Xie, Y., Williams, N.S., and Nijhawan, D. (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356. 10.1126/science.aal3755.
- Dong, G., Ding, Y., He, S., and Sheng, C. (2021). Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. Journal of Medicinal Chemistry 64, 10606-10620. 10.1021/acs.jmedchem.1c00895.
- Zeng, M., Xiong, Y., Safaee, N., Nowak, R.P., Donovan, K.A., Yuan, C.J., Nabet, B., Gero, T.W., Feru, F., Li, L., et al. (2020). Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem Biol 27, 19-31 e16. 10.1016/j.chembiol.2019.12.006.
- Gui, W., Giardina , S., Balzarini, M., Barany, F., and Kodadek, T. (2022). Self-Assembly of Proteolysis Targeting Chimeras Via Reversible Bioorthogonal Reactions. ChemRxiv. 10.26434/chemrxiv-2022-1g1p4.
- Janse, D.M., Crosas, B., Finley, D., and Church, G.M. (2004). Localization to the proteasome is sufficient for degradation. J Biol Chem 279, 21415-21420. 10.1074/jbc.M402954200.
- Yu, H., Kago, G., Yellman, C.M., and Matouschek, A. (2016). Ubiquitin-like domains can target to the proteasome but proteolysis requires a disordered region. The EMBO Journal 35, 1522-1536. 10.15252/embj.201593147.
- Takeuchi, J., Chen, H., and Coffino, P. (2007). Proteasome substrate degradation requires association plus extended peptide. Embo J. 26, 123-131.
- Bashore, C., Prakash, S., Johnson, M.C., Conrad, R.J., Kekessie, I.A., Scales, S.J., Ishisoko, N., Kleinheinz, T., Liu, P.S., Popovych, N., et al. (2023). Targeted degradation via direct 26S proteasome recruitment. Nature Chemical Biology 19, 55-63. 10.1038/s41589-022-01218-w.3
- Balzarini, M., Gui, W., Jayalanth, I.M., Tong, J., Schell, B.-B., and Kodadek, T. (2023). Chemically induced degradation of native proteins by direct recruitment to the 26S proteasome. J. Biol. Chem. 299, Submitted.
- King, E.A., Cho, Y., Dovala, D., McKenna, J.M., Tallarico, J.A., Schirle, M., and Nomura, D.K. (2022). Chemoproteomics-Enabled Discovery of a Covalent Molecular Glue Degrader Targeting NF-κB. bioRxiv, 2022.2005.2018.492542. 10.1101/2022.05.18.492542.
- Peth, A., Kukushkin, N., Bosse, M., and Goldberg, A.L. (2013). Ubiquitinated proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. J Biol Chem 288, 7781-7790. 10.1074/jbc.M112.441907.
- Peth, A., Besche, H.C., and Goldberg, A.L. (2009). Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol Cell 36, 794-804. 10.1016/j.molcel.2009.11.015.
About The Authors:
 Weijun Gui is postdoctoral scholar in the laboratory of Prof. Thomas Kodadek in the Department of Chemistry at The Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology. He received his B.A. in chemistry from Wuhan Textile University and his Ph.D. in organic chemistry from the University of Delaware in the laboratory of Prof. Zhihao Zhuang. He is interested in chemical biology, development of new strategies for targeted protein degradation, and the manipulation of protein homeostasis with small molecules.
Weijun Gui is postdoctoral scholar in the laboratory of Prof. Thomas Kodadek in the Department of Chemistry at The Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology. He received his B.A. in chemistry from Wuhan Textile University and his Ph.D. in organic chemistry from the University of Delaware in the laboratory of Prof. Zhihao Zhuang. He is interested in chemical biology, development of new strategies for targeted protein degradation, and the manipulation of protein homeostasis with small molecules.
 Thomas Kodadek is a professor of chemistry at the Herbert Wertheim UF Scripps Institute for Biomedical Research & Innovation (formerly the Florida campus of the Scripps Research Institute). He received his B.S. in chemistry from the University of Miami (1981), his Ph.D. in organic chemistry from Stanford University (1985) and conducted postdoctoral studies with Prof. Bruce Alberts at UCSF (1985–1987) focusing on DNA replication enzymology. He is an expert in the field of chemical biology with more than 250 publications. Kodadek is a co-founder of Deluge Biotechnologies and Triana Biomedicines and sits on the Scientific Advisory Board of several companies. He can be reached by email at kodadek@ufl.edu.
Thomas Kodadek is a professor of chemistry at the Herbert Wertheim UF Scripps Institute for Biomedical Research & Innovation (formerly the Florida campus of the Scripps Research Institute). He received his B.S. in chemistry from the University of Miami (1981), his Ph.D. in organic chemistry from Stanford University (1985) and conducted postdoctoral studies with Prof. Bruce Alberts at UCSF (1985–1987) focusing on DNA replication enzymology. He is an expert in the field of chemical biology with more than 250 publications. Kodadek is a co-founder of Deluge Biotechnologies and Triana Biomedicines and sits on the Scientific Advisory Board of several companies. He can be reached by email at kodadek@ufl.edu.