
Replacing The MAC/MACO With The MSC: Rethinking How Cleaning Validation Limits Are Calculated
By Andrew Walsh; Thomas Altmann; Ralph Basile; Joel Bercu, Ph.D.; Alfredo Canhoto, Ph.D.; Andreas Flueckiger, M.D.; Igor Gorsky; Jessica Graham, Ph.D.; Ester Lovsin Barle, Ph.D.; Ovais Mohammad; Mariann Neverovitch; Siegfried Schmitt, Ph.D.; and Osamu Shirokizawa

This article discusses two of the most commonly used terms used in cleaning validation: the Maximum Allowable Carryover (MAC or MACO) and the Maximum Safe Carryover (MSC). Both the MAC and MSC are the initial values calculated in the overall calculation of cleaning validation acceptance limits for each individual swab or rinse samples or for a visual inspection. In this article we will demonstrate how only the MSC embraces the risk-based approach to cleaning validation demanded by regulatory guidelines and standards. Below, we provide the historical development of both terms and the technical basis for this position.
The term MAC has been in widespread use in the pharmaceutical industry for many years. In contrast, the term MSC originated with the 2018 publication of the ASTM E3106 Standard Guide.1 While these terms appear the same and would appear to have similar meanings, they are, in fact, based on very different philosophical concepts. Unfortunately, these terms are now being used interchangeably due to confusion, misconceptions, and misunderstanding of both terms. This article will first review the origin of each term, then attempt to resolve the confusion associated with the terms and, finally, present the rationale for use of the MSC.
This article will then address the following four questions:
- Is the Maximum Allowable Carryover safe?
- Is the Maximum Safe Carryover safe?
- Is the Maximum Allowable Carryover allowable?
- Is the Maximum Safe Carryover allowable?
The Maximum Allowable Carryover
While the maximum allowable carryover and its acronyms (MAC and MACO) are widely used in the industry, the origin of this term is unclear. The early ideas on setting cleaning validation acceptance limits and the origin of the 0.001 dose-based limit have been discussed in another article.2 An article by James Agalloco in 19923 presented nine options for answering the question "How much residual will be permitted?" but did not present the concept of the terms MAC or MACO. The well-known and widely cited Fourman and Mullen article on calculating cleaning acceptance limits from 19934 made no mention of MAC or MACO and only provided some of the steps in the calculation of analytical limits that is currently used. Another article in 1998 described the calculation of acceptance limits broken into three steps; this article did not use either of the terms MAC or MACO but made reference to an LSP (limit in subsequent product). 5
MAC seems to have first appeared in the PDA Technical Report No. 29 (TR 29)6 in 1998 in the Definitions section. TR 29 was developed in large part directly from the Agalloco article. Although the term was not mentioned at all in the text of the 1998 TR 29, MAC is defined as:
"the maximum amount of carryover from one product to the next that will not produce a therapeutic dose, corrected for a safety factor (e.g., 1/1,000)."
The acronym MACO did not appear in the 1999 APIC Guide7 but then does appear in the 2000 version. In the APIC 2000 version, the definition provided for MACO is:
"acceptable transferred amount from the investigated product ("previous")."
While both MAC and MACO are terms widely used in the industry, it is probably safe to say that neither of these definitions are currently used by most companies. Most companies would probably define the MAC or MACO in similar but in different ways, if they define them at all.
The MAC, or MACO, is widely being calculated in the industry as follows:

The Maximum Safe Carryover (MSC)
Similar to MAC/MACO, the specific origin of the MSC is not exactly clear from the literature and has its origin in a completely different term – the Safe Threshold Value (STV). The term STV itself grew organically during the development of the ISPE Risk-MaPP (Risk-Based Manufacture of Pharmaceutical Products) Guide8 from two other terms: Safe Limit and Safety Threshold, both coined by one of the authors of this article (Walsh).
Both the Safe Limit and Safety Threshold first appeared in presentations given to the FDA and at ISPE conferences during the development of the Risk-MaPP Guide. As a follow-up to a June 2005 ISPE Conference, Edwin Melendez (FDA, national penicillin expert) requested several speakers to provide information to FDA at its Rockville, MD, headquarters in January of 2006. At this training session, Safe Limit was used by Andrew Walsh in his presentation.9 Then in an ISPE webcast in September 2007, Safety Threshold was introduced as a value "derived from the ADI" in a presentation by Andrew Walsh.10 The same term was used in a presentation to FDA in November of 2007, also by Andrew Walsh.11 Safety Threshold was also used in a presentation to NJ-ISPE members in November 2007.12 The same term was again used in an ISPE presentation by Andrew Walsh in Washington, D.C., in June of 2008.13
During this time, these terms were considered by Walsh to be somewhat confusing, and their significance was not exactly clear based on questions received from some attendees of these presentations. Walsh requested input from the members of the Risk-MaPP team in coming up with a more relevant and understandable term; however, no replacement was suggested prior to the publication of Risk-MaPP in 2010. At the time of publication of Version 1 of the Risk-MaPP Guide, a new term, Safe Threshold Value, (STV) was introduced and its definition was also deemed to be confusing and inadequate:
"The Safe Threshold Value is calculated from the Acceptable Daily Exposure and is the upper limit for statistical analysis used to determine the process capability and cleaning validation limits."
During the development of the ASTM E3106 Standard,1 a new term was introduced by the ASTM Cleaning Team, the Maximum Safe Carryover, whose purpose was to provide a clearer and more concise definition than the MAC, as well as to distinguish itself from the MAC. The term MSC was first introduced in an article in 201114 as a replacement for the MAC/MACO when calculating a cleaning limit using the acceptable daily exposure (ADE). The MSC was not specifically defined in the article, and it was only noted that the MSC is "...equivalent to the term STV found in Risk-MaPP Version 1."
A definition for the MSC was provided in the ASTM E3106 Standard published in 2017 as the:
"maximum amount of carryover of a residual process residue (API, cleaning agent, degradant, and so forth) into the next product manufactured without presenting an appreciable health risk to patients." (emphasis added)
This definition was meant to make it clear that the calculated carryover did not present a health risk to a patient and this amount of carryover is safe. However, this definition did not make a claim that this amount of carryover is allowable in any way.
Subsequent to this, in Version 2 of the Risk-MaPP Guide (also published in 2017), Safe Threshold Value was not used in the text at all and its definition was also removed. There was no explanation offered for its removal nor was any replacement for it suggested.
The preceding narrative might demand an explanation and clarification on its own, but the need for this article was more recently underscored by the release of the Health Canada Guidance 0028,15 which uses the terms Maximum Allowable Carryover, Maximum Safe Carryover and the Safe Threshold Value within the same guidance. Also included in this guidance are two new terms, "maximum safe carryover limit" and "safe daily threshold value", further confusing all of these terms. No definitions for any of these terms were provided in the guidance, which suggests that even regulators are unclear under which terms the industry should be operating.
The MSC is calculated as follows:

Note that the only difference between Equation 2 and Equation 1 is that the Health-Based Exposure Limit (HBEL) replaces the LTD/SF. All the subsequent calculations used for analytical limits (swab/rinse limits) are the same as those used for the MAC.
With this historical background reviewed, we would like to discuss the four questions posed in the introduction.
Is The Maximum Allowable Carryover SAFE?
NO, not always.
This disturbing answer was first discussed in an article in 2011,14 where it was pointed out that the exposure limits based on therapeutic doses are not related to the hazard levels of drugs and may not adequately provide patient safety if the contaminants have characteristics such as being teratogenic, genotoxic/carcinogenic, or sensitizing. To demonstrate how severe this discrepancy could be, an example was shown where a low hazard drug (low-dose aspirin) would have lower limits than a more hazardous chemotherapy agent (capecitabine). The article then called for using the ADE as the starting point for calculating cleaning validation limits to ensure that the cleaning limits were safe for all subsequent products.
Just prior to this article, in 2010, the Risk-MaPP Guide had introduced the use of the ADE as the starting point for cleaning validation limit calculations. In the Risk-MaPP Guide, the ADE was defined as:
"A dose that is unlikely to cause an adverse effect if an individual is exposed, by any route, at or below this dose every day for a lifetime."
This is an extremely stringent (severe) limit – a level of drug that a patient could be exposed to every day for their entire life – with no expectation of adverse effects. So, while the ADE would clearly provide for very safe limits for patient exposure, it raised concerns in the industry at the time that many previously validated cleaning processes might now fail this limit. Many companies resisted implementing the ADE, complaining that this may require extensive revalidation efforts. However, this would have only required companies to revisit their older calculations that used the non-health based 0.001 dose and arbitrary 10 ppm value, which is no more than a simple spreadsheet exercise. There was also resistance against the need to derive documented HBELs for every API made in a shared facility without even taking into consideration other contaminants, such as excipients, cleaning agents, and degradants.
Then in November of 2014, the European Medicines Agency (EMA) issued a guideline on HBELs,14 providing guidance to the hazard assessment experts on how to set HBELs. The EMA defined the HBEL similarly to the Permitted Daily Exposure (PDE) level as:
"...a substance-specific dose that is unlikely to cause an adverse effect if an individual is exposed at or below this dose every day for a lifetime."
Note that in this guideline the EMA recognized the ADE and the PDE to be equivalent.
In 2016, a study was published that compared 304 HBEL (ADE) values against the 0.001 dose-based limits from several large pharmaceutical companies.17 This study revealed situations where the previous 0.001 dose-based limits were not low enough and potentially posed a risk to patients. The study also revealed situations where the previous the 0.001 dose-based limits were lower (often much lower) than those calculated using the HBEL.
Figure 1 shows a comparison of the HBEL values vs. 0.001 of the lowest clinical dose for these 304 pharmaceutical products. Many of the differences between these two values were so great that in order to show them all on one graph they had to be plotted as the ratios of the two values on a semi logarithmic plot. These ratios showed that, in about 15% of the cases, the HBEL was lower than the 0.001 dose, including two instances in which the HBEL was 100 times lower, indicating a potential concern for patient safety. On the other hand, these ratios also showed that in 85% of the cases, the HBEL was 10 to 100 times higher than the 0.001 dose limit, demonstrating that, for many drugs, the 0.001 dose limit has been overly conservative by a very large margin (10X to 100X).
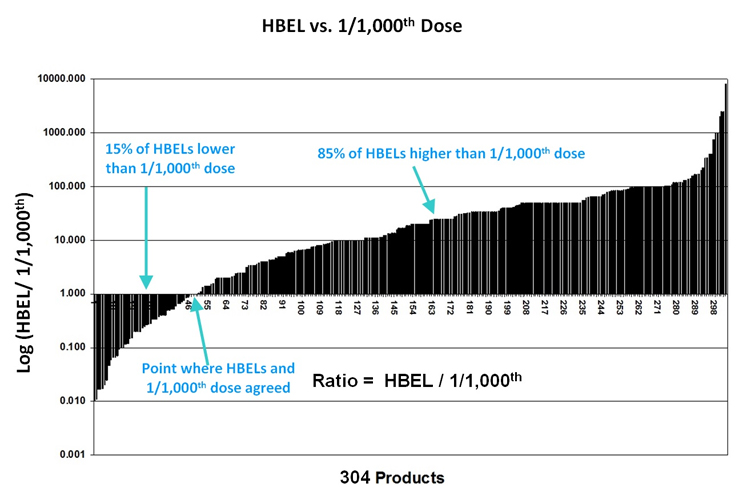
Figure 1 - Comparison of a Compound's ADE to its Corresponding 1/1,000th (0.001) Dose. The data were plotted as a ratio of the HBEL to the 0.001 dose, which makes 1.00 on the Y axis the point at which the HBEL equals the 0.001 dose (Note: the ratios had to be plotted on a logarithmic scale in order to get all the data on one graph because the values of the ratios varied widely). (Graph created by Ester Lovsin-Barle, Ph.D., and used with permission from Walsh, et.al.15 Note: the graph has been updated from the original article replacing the term ADE with the HBEL.)
MAC values have also been calculated for these 304 products using both the 0.001 dose and 10 ppm limit and plotted with the corresponding MSC value based on their HBELs. These MAC value comparisons are shown in Figure 2.
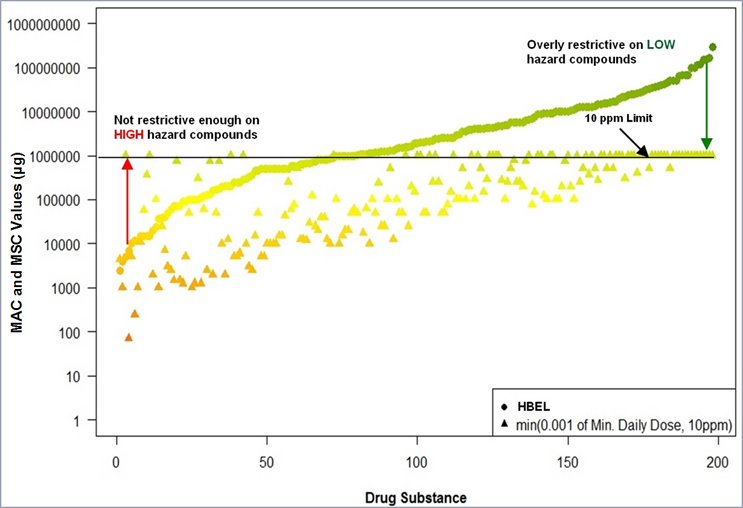
Figure 2 - Comparison of MSC values calculated from HBELs with MAC values calculated from 0.001 dose and 10 ppm. Graph created in R by Ovais Mohammad.
The left side of the graph in Figure 2 shows that there are several compounds where the MACs calculated using the 0.001 Min. Daily Dose (∆) are much higher (10X to 100X) than the MSCs calculated using their HBELs (●) and this is where the 0.001 Min. Daily Dose approach is not restrictive enough on some more hazardous compounds. There are also four compounds where the alternative 10 ppm limit was imposed, and these were still significantly higher than the HBEL based MSCs.
Based on this evaluation, the MAC cannot be considered safe for all product changeover situations as it is not based on an actual science.
Is The Maximum Safe Carryover SAFE?
YES. By definition.
The MSC is derived directly from the HBEL and, therefore, patients are expected to be protected if they are exposed at or below this dose every day for a lifetime. The basis of the MSC calculation is performed specifically to find what level of carryover is safe based on health-based data. It is important to understand and remember that the HBEL (calculated by Qualified Experts) is, by its own definition, an extremely stringent limit.
Is The Maximum Allowable Carryover ALLOWABLE?
NO.
This answer is primarily based on the fact that the MAC is NOT always safe in the first place, as discussed earlier. This was also discussed in the article in 2011 where hypothetical MAC values were calculated for two hypothetical products using their low clinical doses, large and small batch sizes, and high and low maximum daily doses. All possible combinations of these factors (23) were calculated and the data then sorted by their MAC values from lowest to highest (Table 1). Although some of these combinations would not actually occur in a manufacturing operation, this simulation is meant to demonstrate how widely varying the MAC can be.

Table 1 - Comparison of Hypothetical MAC Values. The table shows all possible combinations of lowest dose (Low = 0.001 mg / High = 500 mg), batch size (Low = 15 kg / High = 1,200 kg) and maximum daily dose (Low = 0.05 gm / High = 5 gm) for a hypothetical facility. Table modified and used with permission from Walsh.12
In some cases, merely cleaning to these calculated limits would leave large amounts of residues and in other cases the calculated limits could be below analytical capabilities and impossible to meet. This analysis demonstrates that MAC values can vary by many orders of magnitude for the same drug compound depending on the batch sizes and maximum daily doses of the next drug product. It should be obvious that the MAC calculation can result in excessive amounts of carryover that would never be allowable in a GMP environment.
This problem did not go unnoticed by FDA and was raised as a serious GMP concern at an internal FDA presentation as far back as 2001.18 At this presentation it was said:
"The concern relates to specific recommendations that are being suggested for determining cleaning limits and acceptance criteria for product to product carry-over known as "Maximum Allowable Carryover" or MAC limits. The recommendations are based on mathematical formulas that allow a certain fraction of the therapeutic dose to carry over into each dosage unit of the following product. The intent of this paper is to bring to light the serious flaws inherent in this recommendation and to demonstrate that the mathematical formulas themselves cannot reliably be used to establish acceptance criteria for equipment cleaning." (emphasis added by authors)
The message in this presentation was that just because the industry can do a calculation that yields a high limit does not mean that this is now an "allowable" carryover within the GMPs or to an inspector. Essentially, no cross contamination should be allowable if one can easily prevent it – the goal should always be to minimize cross contamination and potential adulteration of the next product and not "allow" for it.
So, the MAC was already not considered allowable over 20 years ago by some FDA inspectors and it should definitely not be considered allowable today as, once again, it is not based on patient safety (i.e., understanding of drug safety).
Is The Maximum Safe Carryover ALLOWABLE?
NO.
The MSC is not "allowable" for the very same reasons as discussed in the answer to "Is the Maximum Allowable Carryover ALLOWABLE?" above. This was also discussed in the 2011 article, where the same analyses for the Maximum Allowable Carryover were also applied to the Maximum Safe Carryover. So, while the MSC may be "safe," it should not be considered "allowable," either.
Discussion
There have been ongoing discussions, and arguments at times, around how to set limits for the cleaning of pharmaceutical equipment for many years. One of the earliest, if not the earliest, article discussing cleaning limits was published by Sam Harder in 1984.19 In his article, Harder asserted that:
- the limits should be practical and achievable by a reasonable cleaning procedure
- the limits must be verifiable by analytical methodology existing at the company
- the limits must be safe and acceptable and in line with residual limits set for various substances in food.
Many cleaning validation practitioners will immediately recognize the phrase "practical, achievable, and verifiable," which has appeared in many articles, conference presentations, company cleaning validation policies, and master plans, including the FDA's Guide to Inspections of Cleaning Processes. But cleaning validation practitioners will probably be surprised to find out that the "safe and acceptable" requirements were dropped in the FDA's guide and, soon after, everywhere else. Why "safe and acceptable" were not included is not clear. The reason for this omission may never be known as the principal author, Henry (Hank) Avallone, passed away in 2008.
While there are a few people in the cleaning validation community who have claimed that the use of the 1/1,000 dose criteria was "settled" after FDA mentioned them in their 1993 guide, this cannot be further from the truth.
The FDA's 1993 guide did not specifically endorse the use of the 1/1,000 dose criterion. The guide stated:
"Some limits that have been mentioned by industry representatives in the literature or in presentations include analytical detection levels such as 10 PPM, biological activity levels such as 1/1,000 of the normal therapeutic dose, and organoleptic levels such as no visible residue."
This statement only provided the inspector with some limits that FDA was aware of in 1993 based on publications and presentations at that time, and this does not signify overt acceptance at all. However, it should be noted that the adjective “scientifically justifiable” was included with the word “limits,” which implied that each manufacturer should be able to scientifically defend how they selected their limits. The Guide states:
"The objective of the inspection is to ensure that the basis for any limits is scientifically justifiable."
So, the FDA's guide clearly instructs the inspectors to look for a scientific justification for the limits used and not to just accept these limits mentioned in the guide without question.
More recently, in 2015 FDA issued a Q&A20 where it states,
"Equipment should be as clean as can be reasonably achieved to a residue limit that is documented to be safe, causes no product quality concerns, and leaves no visible residues. Contamination that is reasonably avoidable and removable is never considered acceptable."
These statements can probably trace their origins to contemporary quality initiatives such as the 2006 ICH Q9 Guidance with its focus on patient safety, the 2010 Risk-MaPP guidance with its ADE concept and the EMA's 2014 HBEL Guideline with its PDE concept.
It should be understood that not only has the MAC never been 100% "safe," it has also never truly been "allowable." While the PDE-based calculation is truly "safe" now, it can also result in carryover levels that should not be "allowable," either. That is why the MSC was developed for use with the HBEL (PDE/ADE). While the MSC should be considered "safe," it should not be considered "allowable."
It is hoped and firmly encouraged that the industry will move away from the use of the term MAC and begin using MSC and continue progressing toward truly science- and risk-based cleaning validation. In addition, the MSC should be seen only as a tool for measuring the level of risk of a compound14 and that control limits should be calculated from the data collected during cleaning activities using statistical process control (SPC) techniques14 (see Figure 3), as is expected in 21 CFR 211, FDA cGMPs, and FDA's Process Validation Guidance,21 as well as EMA Guidance.22 These SPC limits would then be used as acceptance limits for future cleaning qualifications or verifications.
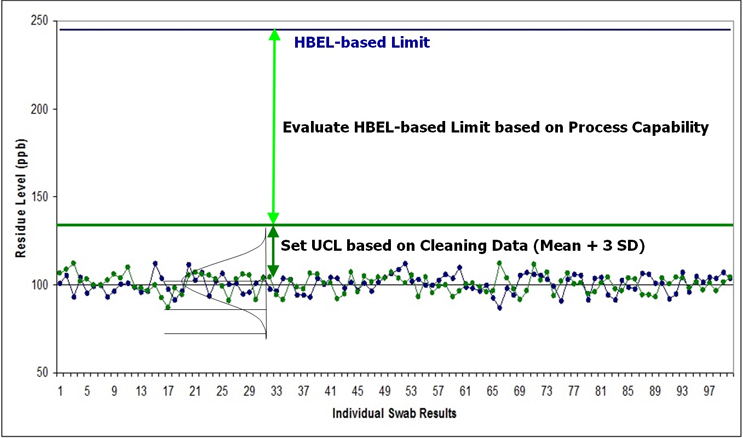
Figure 3 - Process Capability Measured by HBEL-based Limits and Statistical Process Control Limits Calculated from Cleaning Data. The cleaning data collected during cleaning process development or qualification runs are compared to the HBEL-based limit to determine the process capability of the cleaning process. The process capability determines the level of risk for the cleaning process and may initiate risk reduction activities. After the cleaning process development and qualification runs are completed, the collected data are used to set a cleaning process control limit using SPC techniques. Such SPC-derived cleaning process control limits are "data-derived" and reflect what the cleaning process is capable of achieving. Cleaning data collected in all subsequent cleaning validation or verification runs would then be evaluated using these cleaning "data-derived" limits. Note: such SPC limits may be updated when new data is available.
Peer Review
The authors wish to thank James Bergum, Ph.D.; Gabriela Cruz, Ph.D.; Mallory DeGennaro; Christophe Gamblin; Ioanna-Maria Gerostathes; Ioana Gheorghiev, M.D.; Jessica Graham, Ph.D., DABT; Hongyang Li; Ajay Kumar Raghuwanshi; and Ersa Yuliza for reviewing this article and for providing insightful comments and helpful suggestions.
References
- American Society for Testing and Materials (ASTM) E3106-18 "Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation" www.astm.org.
- Walsh, Andrew “Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part I” Pharmaceutical Engineering, July/August 2011, Volume 31, Number 4, pp. 74-83, www.ispe.org.
- Agalloco, James "Points to Consider in the Validation of Equipment Cleaning Procedures" Journal of Parenteral Science & Technology Vol 46, No. 5 / September-October 1992
- G. Fourman and M. Mullin “Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations” in “Pharmaceutical Technology” April 1993.
- LeBlanc, Destin, "Establishing Scientifically Justified Acceptance Criteria for Cleaning Validation of Finished Drug Products" in "Pharmaceutical Technology" October 1998.
- Parenteral Drug Association, Points to Consider for Cleaning Validation, Technical Report No. 29, August 1998.
- Active Pharmaceutical Ingredients Committee (APIC) of European Chemical Industry Council (CEFIC) "Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants" 1999.
- ISPE. (2010). Baseline® Pharmaceutical Engineering Guide: Risk-Based Manufacture of Pharmaceutical Products: A Guide to Managing Risks Associated with Cross-Contamination (First ed. Vol. 7). Tampa, FL: ISPE.
- Training session on Cleaning to FDA in Rockville MD January 2006 "Manufacturing of “Potent Drugs”: An Industry Perspective on Facility Design, Measurements and Controls".
- ISPE Webcast September 2007 "An Introduction to ISPE's Risk-MaPP Baseline Guide and Its Application to All Pharmaceutical Compounds"
- Presentation to FDA November 2007 "Science and Risk Based Approaches To Acceptance Limits".
- Presentation to NJ-ISPE November 2007 "New Perspectives on Cleaning Validation: Science and Risk Based Approaches".
- ISPE Presentation June 2008 "Cleaning Validation Acceptance Limits – Past, Present and Future"
- Walsh, Andrew "Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part II", Pharmaceutical Engineering, September/October 2011. Vol. 31 (No. 5)
- Health Canada: Cleaning Validation Guide (GUI-0028) V5, June 29, 2021
- EMA Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities, EMA/CHMP/CVMP/ SWP/169430/2012, 20 November 2014. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/11/WC500177735.pdf.
- Walsh, Andrew, Michel Crevoisier, Ester Lovsin Barle, Andreas Flueckiger, David G. Dolan, Mohammad Ovais (2016) "Cleaning Limits—Why the 10-ppm and 0.001-Dose Criteria Should be Abandoned, Part II," Pharmaceutical Technology 40 (8)
- DeMarco, Ann L. and Susan Jansen-Varnum, "A Perspective on Equipment Cleaning - Industry practice but NOT cGMP -" Central Atlantic States Association: Millennium Program for the Pharmaceutical Industry May 15, 2001
- Harder, S.W. "The validation of cleaning procedures" Pharmaceutical Technology, May 1984
- https://www.fda.gov/drugs/guidances-drugs/questions-and-answers-current-good-manufacturing-practices-equipment#5
- Guidance for Industry Process Validation: General Principles and Practices, January 2011
- April 2018 EMA/CHMP/CVMP/SWP/246844/2018 Questions and answers on implementation of risk-based prevention of cross-contamination in production and ‘Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities’ (EMA/CHMP/CVMP/SWP/169430/2012)