
Revision Of The ASTM E3263 Standard For Visual Inspection Of Pharmaceutical Manufacturing Equipment And Medical Devices For Residues
By Andrew Walsh; Thomas Altmann; Ralph Basile; Stéphane Cousin; Delane Dale; Parth Desai; Boopathy Dhanapal; Jayen Diyora; Christophe Gamblin; Igor Gorsky; Jove Graham; Reto Luginbuehl; Spiro Megremis; Ovais Mohammad; Mariann Neverovitch; Laurence O'Leary; Rod Parker; Siegfried Schmitt, Ph.D.; Osamu Shirokizawa; Stephen Spiegelberg, Ph.D.; and Norma Turner

Part of the Cleaning Validation For The 21st Century Series
ASTM has published the revision of ASTM E3263 "Standard Practice For Qualification Of Visual Inspection Of Pharmaceutical Manufacturing Equipment And Medical Devices For Residues.” Why has a revision been published only a short two years after the 2020 version of the Standard?
Back in April of 2018, the European Medicines Agency (EMA) had posted a Q&A on their guideline for setting health-based exposure limits.1 In it, two new questions and answers appeared (Q#7 and Q#8) that are directly applicable to the use of visual inspection in cleaning. In these two Q&As, the regulatory requirements for implementing visual inspection as one of the tools available for cleaning validation were now rather well defined and all that was needed now was a detailed guidance on how to implement cleaning programs that satisfy these criteria.
In March of 2019, members of the ASTM E55 Committee on Manufacture of Pharmaceutical and Biopharmaceutical Products and members of the ASTM F04 Committee on Medical and Surgical Materials and Devices collaborated on writing a new Standard Guide on Visual Inspection that resulted in the publication of ASTM E3263-20 in December 2020.2
The ASTM E3263-20 had been written, in part, to provide the necessary guidance for establishing qualified visual inspection programs to comply with these newly clarified regulatory expectations. A detailed article introducing the standard was simultaneously published on Pharmaceutical Online, Bioprocess Online, and Med Device Online3 and these articles were announced in social media discussion groups involved with pharmaceutical and medical device cleaning. Shortly after announcing these articles, the article authorship team was contacted by a former inspector with the Medicines and Healthcare Products Regulatory Agency (MHRA) to obtain a copy of this standard, which was happily provided by ASTM. After his review, the former MHRA inspector asked the team if it was possible to make some revisions to make the standard more acceptable to the Pharmaceutical Inspection Co-Operation Scheme (PIC/S), which had recently adopted the European Medicines Agency’s (EMA) Q&A on Health Based Exposure Limits.4
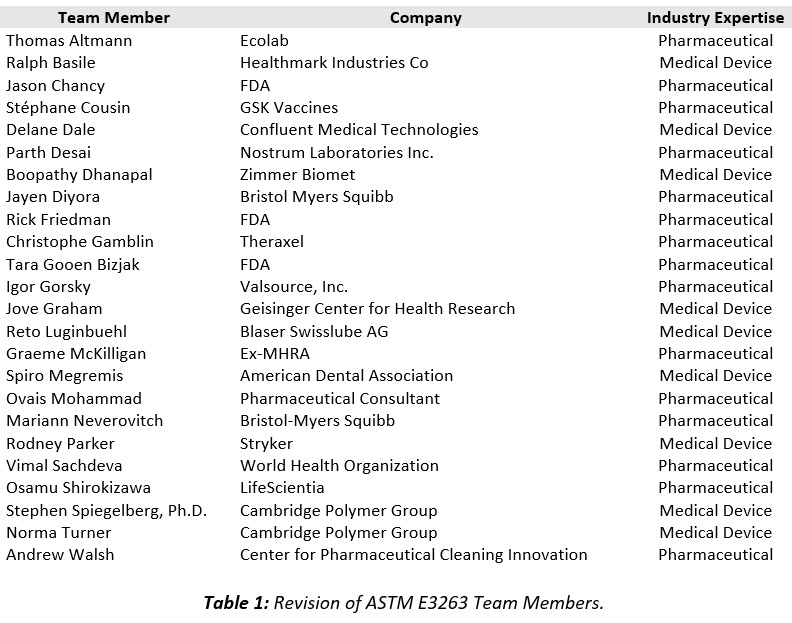
The E55/F04 Joint Cleaning Team immediately initiated a new Work Item on the ASTM website and reinstated their weekly meetings to revise the ASTM E3263 with the participation of the former MHRA inspector, and with the addition of a WHO inspector and three representatives of the FDA. The full team that participated in the revision of ASTM E3263 is shown in Table 1.
This revision was balloted in ASTM, approved, and is now available on the ASTM website (https://www.astm.org/e3263-22.html). Instead of the rest of this article being a full summary of the newly revised E3263 Standard, we focus on its updates compared to the 2020 Standard.
Scope Of ASTM E3263 Revision
The E3263 provides statistically valid procedures for determining the visual detection limit of residues and the qualification of inspectors to perform the visual inspection of pharmaceutical manufacturing equipment surfaces and medical devices for residues.
E3263 applies to:
- Pharmaceuticals, which includes active pharmaceutical ingredients (APIs); dosage forms; and over the counter, veterinary, biologics, and clinical supplies. This practice is also understood to be applicable to other health, cosmetics, and consumer products.
- Chemical Residues, including degradants, intermediates, cleaning agents, processing aids, machining oils, etc., that could remain on manufacturing equipment surfaces or medical devices that have undergone all manufacturing steps including cleaning.
- Equipment and Devices that have been justified through a quality risk management program with an acceptable hazard analysis, have cleaning validation processes that are repeatable and validated, and visual inspection that has been qualified and can be relied upon to determine the cleanliness of the equipment at the residue limit justified by the HBEL.
Significance And Use Of ASTM E3263
The significance and use of the E3263 are to specifically provide instructions for qualification of visual inspection for residues on pharmaceutical manufacturing equipment and medical devices. It provides guidance on the following:
- An approach that applies the science-based, risk-based, and statistics-based concepts and principles introduced in Guides E31065 and E32196
- An approach for qualifying the (visual) inspection of equipment for cleanliness in accordance with 21 CFR 211.67(b)7 and is in accordance with the FDA Process Validation Guidance Life Cycle approach8
- An approach for qualifying the visual inspection of equipment for cleanliness in accordance with European Medicines Agency (EMA) Annex 159
- An approach for qualifying the visual inspection (and visual threshold) of equipment for cleanliness in accordance with the EMA’s Q&A Guidance (Q&A #7 and Q&A #8)3
- Visual Inspection used as described in No. 4 should only be used in situations where there is a suitable safety margin between the visual residue limit (VRL) and the maximum safe surface residue (MSSR) and robust detectability at the VRL.
- An approach that applies the risk-based principles introduced in ICH Q910 so that the level of effort, formality, and documentation for cleaning validation would also be commensurate with the level of risk
- An approach for releasing manufacturing equipment and manufactured medical devices or cleanliness that is compatible with the U.S. FDA’s guidance on its Process Analytical Technology Initiative11
The E3263 Standard Practice Guide
The Procedure section of E3263 contains guidance on six main elements. The first element discusses what initial criteria must be met in order to implement a visual inspection program.
1. Initial Criteria for Establishing Qualification Programs for Visual Inspection:
There are four criteria that need to be met before a visual inspection (VI) program can be considered, and these are derived directly from the EMA guidance.3
- The compounds/products/residues selected for evaluation of VI shall have acceptable hazard levels based on their HBELs derived by a qualified expert.
- The cleaning processes of the compounds/products/residues selected should be validated and not present any significant concerns for patient safety if visual inspection will be used for release of equipment with no additional analytical testing.
- The hazard level of a compound/product and the acceptability of the cleaning process should be evaluated to determine acceptability using a risk management tool such as the Shirokizawa Matrix12.
- The VI data collected for these compounds/products/residues shall demonstrate that VI can be relied on for determining the cleanliness of the equipment at the residue limit(s) justified by the HBEL.
If the four initial criteria can be met, there are then six criteria that also need to be met when establishing a VI program.
- Calculation of MSSR – MSSRs must first be calculated for all equipment to be inspected, as it is necessary to determine the minimum level of residue that must be detectable by the visual inspection. The MSSR, expressed in mass units per surface area (for example, μg/cm2), is calculated using the following equation (from ASTM E31065):
MSSR = MSC
TSA
Where:
MSC = Maximum Safe Carryover
TSA = Total Surface Area (of shared equipment or medical device)
- Selection of Surfaces for the Qualification Study – This criterion details the steps to take in selecting the materials of construction for visual inspection studies.
- Selection of Products/Compounds for the Qualification Study – This criterion details the steps to take in selecting the compounds/products (e.g., APIs, cleaning agents, machining oils, etc.) for visual inspection studies.
- Preparation of Surrogate Surfaces or Devices – This criterion details how to prepare the surrogate surfaces (e.g., coupons, equipment parts, medical devices, etc.) for use in visual inspection studies.
- Surrogate Surface Storage and Handling – This criterion details how the surrogates’ surfaces should be handled.
- Viewing (Lighting) Conditions – This criterion details the lighting requirements for performing the visual inspection studies.
2. Inspector Training – This section discusses what is necessary to train operators/QA inspectors for visual inspection and maintain their qualified state.
3. Determination of Visual Residue Limits (VRL) – This section discusses how to identify the lowest spiked residue level (visual threshold) that is most likely to be seen by all trained operators/QA inspectors for the product/compound of a spiked coupon study. This spiked residue level (visual threshold) should be the starting point for inspector qualification studies. In cases where the VRL is determined in a study using a small number of inspectors (N<5), the VRL may not be statistically justifiable. A method for setting scientifically and statistically justifiable VRLs using logistic regression analysis (Figure 1) was developed by Ovais Mohammad to provide a meaningful determination of the VRL.13

4. Qualification of Inspectors Using Attribute Agreement Analysis – This section discusses how to set up a visual inspection study to qualify any number of operators/QA inspectors by analyzing inspection results as attribute/binary data (i.e., clean/dirty).
5. Acceptance of the VRL for Cleaning Validation – This section discusses how to determine whether visual inspection is appropriate for use by comparison to the MSSR using the visual detection index (VDI).14 An important change in the E3263 was the added requirement that the VRL should be at least 10 times lower than the MSSR, giving a VDI of -1.

6. Documentation and Lifecycle Risk Management – This section discusses what key documents are necessary for establishing a visual inspection program. This includes, for example, the initial qualification report of the inspectors, the training records of the inspectors, etc.
7. Data Integrity in the Qualification of Visual Inspection - This new section added requirements for ensuring data integrity and recommendations for collection, handling, and storage of qualification data.
Summary
While the EMA's Q&A #7 and Q&A #8,1 published in 2018, may have been a surprise for many in the industry, they were added to allow companies with products found to be low risk (based on their HBELs and well developed and successfully validated cleaning procedures) the option of using visual inspection at product changeovers.
At the same time, it has been an industry habit for many years to move to visual inspection only for release of equipment by simply stopping swab/rinse testing without any QRM program in place, without any adequate justification, or without any qualification of their operators/QA inspectors. These are unacceptable practices that will inevitably lead to regulatory action resulting in significant costs and reputational damage for these companies. Implementing the procedures as described in this revision to E3263 should prevent this. However, E3263 cannot be implemented completely independent of the ASTM E3106 and E3219 standards and must be coordinated with these guides.
Visual inspection alone will most likely find initial use in cleaning process validations on multi-use equipment for new low-risk products. At least initially, this will require that prior satisfactory validation studies of the cleaning process already exist. The six-step QRM process described below would be an appropriate procedure to implement visual inspection.
- The HBEL of the new product must be derived by a qualified expert and compared to the HBELs of the existing portfolio of products. A recommended approach is to use the toxicity scale.15 If the hazard (toxicity) level is acceptable, the product can move to step 2.
- HBEL-derived cleaning (swab) limits should be calculated for the new product, compared to the existing cleaning data for the equipment, and the potential process capability score (Cpu score) of the cleaning process calculated.16 If the Cpu Score is acceptable, the product can move to step 3.
- The "cleanability" of the new product is measured and compared to the existing "hardest-to-clean" product.17 If the cleanability of the new product is acceptable, the product can move to step 4.
- The visual detection limit (visual threshold) should be determined for the new product and the VDI calculated.14 If the VDI is acceptable, visual inspection alone could be justified.
- All operators/QA inspectors are qualified to visually inspect the new product.
- Visual inspection of 100 percent of (disassembled) equipment surfaces should be performed and documented after cleaning each batch of the new product.
We believe that the new E3263 standard provides the science-, risk-, and statistics-based guidance and the tools needed for companies to implement the use of visual inspection within a QRM program that meets the criteria promulgated in the EMA's Q&A #7 and Q&A #8. A case study and an analysis of its metadata have already been published.18, 19
However, it should be understood that regulators are highly unlikely to accept visual inspection alone for cleaning validation unless manufacturers have exceptional justification (such as a very low-hazard product) and will still likely expect some analytical testing to confirm acceptable cleaning during the cleaning performance qualification phase. Where visual inspection can most likely be used alone is in subsequent cleaning process validations (verifications) for new low-risk products on multi-use equipment where prior satisfactory validation studies of the cleaning process have already been performed. The revised ASTM E3263 is in complete alignment with this viewpoint.
Moving even further in this direction, PIC/S20 has now stated that:
“...spiking studies should determine the concentration at which most active ingredients are visible.”
This indicates that these health agencies are expecting to see visual inspection being used more frequently as a semi-quantitative tool and have requirements for its use. This statement specifically about active ingredients (APIs) has led some manufacturers to set up visual inspection and the related training activities to focus only on active ingredients. However, any compound that is identified as a hazard in the risk (hazard) Identification step and found to be a risk in the risk analysis step may need to be included in a visual inspection qualification and training program. This is especially true for medical device manufacturing, as cleaning agents or processing aids may be the compounds identified as high risks and not APIs.21,22
Peer Review
The authors wish to thank Joel Bercu, Ph.D., Sarra Boujelben; Gabriela Cruz, Ph.D.; Mallory DeGennaro; Andreas Flueckiger, MD; Ioanna-Maria Gerostathi; Ioana Gheorghiev, MD; Hongyang Li; and Ajay Kumar Raghuwanshi for reviewing this article and for providing insightful comments and helpful suggestions.
References
- European Medicines Agency, Questions and answers on implementation of risk-based prevention of cross-contamination in production and Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities, 19 April 2018, EMA/CHMP/CVMP/SWP/246844/2018.
- American Society for Testing and Materials E3263-20 Standard Practice for Qualification of Visual Inspection of Pharmaceutical Manufacturing Equipment and Medical Devices for Residues, www.astm.org.
- Walsh, Andrew, Ralph Basile, Stéphane Cousin, Mariann Neverovitch, Mohammad Ovais and Osamu Shirokizawa, Introduction To ASTM E3263-20: Standard Practice For Qualification Of Visual Inspection Of Pharmaceutical Manufacturing Equipment And Medical Devices For Residues, Pharmaceutical Online January 2021
- Pharmaceutical Inspection Convention: Pharmaceutical Inspection Co-Operation Scheme PI 053-1 1 June 2020 Questions and answers on implementation of risk-based prevention of cross-contamination in production and Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities, https://picscheme.org/docview/1948
- American Society for Testing and Materials E3106-18e1 Standard Guide for Science Based and Risk Based Cleaning Process Development and Validation, www.astm.org.
- American Society for Testing and Materials E3219-20 Standard Guide for Derivation of Health Based Exposure Limits (HBELs), www.astm.org.
- 21 CFR 211.67 Equipment Cleaning and Maintenance
- FDA Guidance for Industry: Process Validation - General Principles and Practices January 2011, U.S. Food and Drug Administration (FDA), www.fda.gov.
- EudraLex Volume 4 Guidelines for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonized Tripartite Guideline, Quality Risk Management – Q9, Step 4, 9 November 2005, www.ich.org.
- FDA Guidance for Industry: PAT – A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance, September 2004, U.S. Food and Drug Administration (FDA), www.fda.gov.
- Walsh, Andrew, Thomas Altmann, Ralph Basile, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G. Dolan Ph.D., Pernille Damkjaer, Andreas Flueckiger, M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle, Ph.D., Ovais Mohammad, Mariann Neverovitch, Siegfried Schmitt, Ph.D., and Osamu Shirokizawa The Shirokizawa Matrix: Determining the Level of Effort, Formality and Documentation in Cleaning Validation, Pharmaceutical Online, December 2019.
- Ovais, M., Statistically Justifiable Visible Residue Limits, Pharmaceutical Technology, 34 (3) 2010.
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron. An MSSR-derived Scale for Assessing the Detectability of Compound-Carryover in Shared Facilities, Pharmaceutical Online December 2017
- Walsh, Andrew, Ester Lovsin Barle, Michel Crevoisier, David G. Dolan, Andreas Flueckiger, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron. An ADE-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities, Pharmaceutical Online May 2017
- Walsh, Andrew, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Robert Kowal, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron. A Process Capability-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities, Pharmaceutical Online August 2017
- Song, Ruijin, Alfredo Canhoto, Ph.D., and Andrew Walsh Cleaning Process Development: Cleanability Testing and "Hardest-To-Clean" Pharmaceutical Products, Pharmaceutical Online January 2019.
- Walsh, Andrew, Dongni (Nina) Liu and Mohammad Ovais, Justification and Qualification Of Visual Inspection For Cleaning Validation In A Low-Risk Multiproduct Facility, Pharmaceutical Online August 2018
- Walsh, Andrew, Dongni (Nina) Liu and Mohammad Ovais, Metadata Analysis - Justification and Qualification Of Visual Inspection For Cleaning Validation In A Low-Risk Multiproduct Facility, Pharmaceutical Online August 2018
- Pharmaceutical Inspection Convention Pharmaceutical Inspection Co-Operation Scheme Guide To Good Manufacturing Practice For Medicinal Products: Annex 15 PS/INF 11/2015 1 April 2015
- Bonsignore LA, Anderson JR, Lee Z, Goldberg VM, Greenfield EM. Adherent lipopolysaccharide inhibits the osseointegration of orthopedic implants by impairing osteoblast differentiation. Bone. 2013 Jan;52(1):93-101. doi: 10.1016/j.bone.2012.09.011.
- Bonsignore LA, Victor M Goldberg, Edward M Greenfield, Machine oil inhibits the osseointegration of orthopaedic implants by impairing osteoblast attachment and spreading, J Orthop Res. 2015 Jul;33(7):979-87. doi: 10.1002/jor.22850.