
Strength Biowaivers For Immediate Release Oral Solid Drug Products: European & American Perspectives
By Bilel Khedir, Opalia Pharma Recordati Group

Developing a drug product with multiple strengths has several advantages: it helps meet individual patients’ needs, offers an advantage for drugs requiring titration, avoids splitting tablets (which is bad for drug product stability),1 and in the case of generic drug products it contributes to improving access to quality medicines worldwide.2 However, for a generic drug product, human testing is usually required to demonstrate equivalence, but waivers can be considered in three main cases: self-evident bioequivalence, biopharmaceutics classification system (BCS) based biowaivers, and strength biowaivers.2
A strength biowaiver is usually claimed for an additional strength when bioequivalence has been established for one strength and BCS biowaiver is not applicable.2 Six factors need to be considered with additional strength biowaivers:2
- The pharmacokinetics of the drug substance
- The similarity of the manufacturing process
- The qualitative composition
- The quantitative composition
- The dissolution profile
- The methodology used to analyze dissolution profiles
The same formulation could obtain a strength biowaiver in the U.S. but not in the EU (and vice versa) due to differences in formulation requirements. The same data set of dissolution studies could yield similar profiles (thus similar drug products) in the U.S. but not similar per the EU standards (and vice versa) due to differences in the choice of the last time point. In this article, I will discuss the EMA and FDA requirements and the associated challenges when claiming a strength biowaiver.
1. Pharmacokinetics Of The Drug Substance
The strength on which the bioequivalence studies are done is determined by the pharmacokinetics (PK) of the drug substance. The definition of linear pharmacokinetics and the strength to choose if the PK are linear or nonlinear differ between the EMA and FDA perspectives.
EMA
Linear pharmcokinetics are defined as a dose-adjusted ratio of area under the curve (AUC) of the different strengths within ± 25% (75%–133%).2 The EMA guideline on bioequivalence defines linear pharmacokinetics as occurring when the difference in dose-adjusted mean AUC is no more than 25% when comparing the studied strength (used in the bioequivalence study) and the strength for which a biowaiver is considered.3
Linear pharmacokinetics: In general, BE studies are done on the highest strength, but EMA accepts the lower strength in two cases:
- The drug substance is highly soluble.
- The higher strength cannot be administered for safety/tolerability reasons.
If problems in the sensitivity of the analytical method occur, the administration of a higher dose is tolerated.3
Nonlinear pharmacokinetics: There are two possibilities:
- More than proportional increase in AUC with increasing dose over the therapeutic dose range: Bioequivalence should be established using only the highest strength.
- Less than proportional increase in AUC with increasing the dose over the therapeutic range: In this case, two bioequivalence studies are needed with the highest and lowest strengths.
Exception: If the nonlinearity is caused by saturation of uptake and not limited solubility (the generic and brand products do not contain excipients that may change gastrointestinal motility or transport proteins and the rest of the conditions for the strength biowaiver are fulfilled), it is sufficient to demonstrate the bioequivalence with the lowest strength or a strength in the linear range.3
FDA
The FDA does not provide a definition of linear PK similar to the one given by EMA.1
Linear pharmacokinetics: The FDA recommends conducting the BE study on the highest strength. The use of lower strength is permitted for safety reasons.4
Nonlinear pharmacokinetics: If pharmacokinetics are not linear, the question of the acceptability of a strength biowaiver is an agency review issue unless a product-specific guidance is published that contains details.4
2. Similarity Of Manufacturing Process
The EMA and FDA require the same manufacturing process for all the strengths. Having the same manufacturing process means that both drug products (low and high strength) must have the same pharmaceutical form (if one strength is a tablet, the other must be a tablet, not a capsule, and vice versa). But unlike the FDA, the EMA accepts different manufacturing sites for the strengths of the drug product in question.2
3. Qualitative Composition
EMA and FDA require that all active and inactive ingredients are the same. However, flavors, colors, coatings (other than changes between film and sugar coatings), and inks are allowed to be different.2
4. Quantitative Composition
FDA
The FDA expects that the core of each strength is in the same proportion. On the other hand, for high potency/low percentage (≤10%)2 drug substances, no proportionality is required but three conditions must be fulfilled:
- The total weight of the dosage form remains the same for all strengths (±10% tolerated).
- The same inactive ingredients are used.
- The change in any strength is obtained by altering the amount of the active ingredient and one or more inactive ingredients.
The FDA states that other differences could be accepted with proper justification.1
The FDA also expects each strength to have the same shape.2
EMA1
For the EMA, quantitative composition must be in the same proportion; deviation from this rule is accepted under three conditions:
- The amount of the active substance is less than 5% of the tablet core weight or capsule content.
- The amounts of the different core excipients are the same for the concerned strengths.
- The amount of a filler is changed to account for the active substance change.
5. In Vitro Dissolution Studies
Comparative in vitro dissolution profiles are required by FDA and EMA, but there are some differences in terms of details about:2
- The products to be compared
- The number of units
- The apparatus required
- The media required
FDA2
Products to be compared: Different strengths of test product and comparator (brand) product.
Number of units: 12
Apparatus required: Paddle (50 rpm) and basket (100 rpm) are used.
Media required: The FDA requires testing at the agreed quality control medium. The use of surfcatnat is accepted.
Presentation of dissolution data : A complete set of dissolution profiles
Test product line (generic):
- Reference strength (biobatch : the batch used in the bioequivalence study) (For example, the strength of 100 mg of generic drug product)
- Test strength (For example, 50 mg of generic drug product)
Reference product line (brand):
- Two strengths (100 mg strength of the brand drug and 50 mg strength of the brand drug product)
- The statistical comparison is only required within the test product line, with the biobatch of the reference strength as reference.
EMA2
Products to be compared: The EMA requires dissolution studies to be done within the test product line.
Number of units : 12
Apparatus required: Paddle (50 rpm) and basket (100 rpm) are used.
Media required: Quality control medium and three different pHs over the physiological pH range: 0.1 N hydrochloric acid (pH 1.2), acetate buffer (pH 4.5), and phosphate buffer (pH6.8). The use of surfactant is only possible in the QC medium.
Presentation of dissolution data:
Test product line (generic) :
- Reference strength (biobatch : the batch used in the bioequivalence study) (For example, the strength of 100 mg of generic drug product)
- Test strength (For example, 50 mg of generic drug product)
- Statistical comparison is required between the two strength of the test product line at each medium tested.
Reference product line (brand) :
- Dissolution data of the reference product strengths are only required in some cases, which are discussed in the next section.
6. The Methodology Used To Analyze Dissolution Profiles
Dissolution profiles must be compared by using f2, similarity factor calculation, unless the dissolution of the drug products is very rapid. For the FDA and EMA, very rapid dissolution is when more than 85% of the drug is dissolved for both the reference and the test product within 15 minutes.5
The similarity factor f2 is a statistical parameter used to demonstrate similarity between dissolution profiles. Proposed in 1996 by Moore and Flanner, f2 can be described as the reciprocal square root transformation of the sum of squared error and is a measurement of the similarity in the percent dissolution between two profiles:5
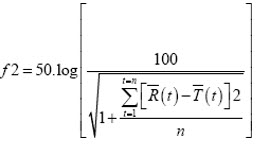
Statistical Analysis5
In order to get reliable results, both agencies (EMA and FDA) impose certain conditions when calculating the similarity factor that cover three main aspects:
- Minimum number of time points
For dissolution profile comparison, the EMA and the FDA require a minimum of three time points. The sampling must be done at least every 15 minutes according to both agencies, but sampling at 5-minute and 10-minute intervals might be useful for rapidly dissolving drug products and very useful to completely chararcterize the dissolution profile and avoid getting a biased f2. Thus, choosing the appropriate sampling times requires a preliminary dissolution study.5
- Last time point
According to the EMA, the last time point to include in f2 analysis is when any one of the reference and the test product has reached 85% or an asymptote is reached. On the other hand, the FDA defines the last time point as the the time point when no more than one mean value (test and reference product) is ˃ 85%.5
For the same data set, applying these two rules might give an f2 of under 50 and more than 50. That’s why the choice of the last time point is very crucial in an f2 analysis.
- Coefficient of variation criteria
The FDA requires a coefficient of variation ≤ 20% for early time points (15 minutes or less) and ≤ 10% at the other time points.5
The EMA requires a coefficient of variation ≤ 20% for the first time point and ≤ 10% from the second to the last time point.5
Interpretation Of In Vitro Dissolution Studies Results
FDA: If the f2 (within test product line strengths) is between 50 and 100, the dissolution profiles are considered similar. However, in the case of strength biowaivers, an f2 of < 50 may be due to the lack of sink conditions. This may be demonstrated by comparing two strengths at the same dose (test product line). The FDA may require brand product line strength f2 calculation on a case by case basis.2
EMA : An f2 between 50 and 100 for dissolution profiles is also required to prove similarity. If the f2 is less than 50, this may be due to the lack of sink conditions; this is demonstrated by comparing two strengths at the same dose (of the test product line). In addition, the EMA requires an f2 < 50 between the two strengths of the brand product line to prove that the difference is drug substance related rather than formulation related.1
Note the definition of sink conditions: The European Pharmacopeia defines sink conditions as dissolution medium volume equivalent to three to 10 times saturation volume. According to the U.S. Pharmacopeia, the sink condition is fulfilled when the dissolution medium is capable of dissolving the amount of drug that is three times greater than the amount of drug to be tested.6
Conclusion
The requirements for strength biowaivers in the EU and the U.S. are different in terms of definition of PK linearity, qualitative and quantitative composition similarity, and the in-vitro dissolution studies required and their statistical analysis (f2 analysis). Furthermore, the choice of a reference product (brand product) is easy from the American perspective, as the FDA has a clear list of reference listed drugs (RLD), while the EU countries might each have different reference drug products.
These differences might require drug companies to formulate a different drug product for each submission, which is very expensive. Thus, a dialogue between the different agencies might be useful to minimize these divergences.
References
- Cardot, J. M., Garcia-Arieta, A., Paixao, P., Tasevska, I., & Davit, B. (2018). Implementing the additional strength biowaiver for generics: EMA recommended approaches and challenges for a U.S. FDA submission. European Journal of Pharmaceutical Sciences, 111, 399-408.
- Crane, C., Santos, G. M. L., Fernandes, E. A. F., Simon, C., Tam, A., Triana, D. G., ... & Garcia-Arieta, A. (2019). The Requirements for Additional Strength Biowaivers for Immediate Release Solid Oral Dosage Forms in International Pharmaceutical Regulators Programme Participating Regulators and Organisations: Differences and Commonalities. Journal of Pharmacy & Pharmaceutical Sciences, 22, 486-500.
- European Medicines Agency (EMA). (2010) Guideline on the investigation of bioequivalence https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf
- Food and Drug Administration (FDA). (2021) Bioequivalence Studies With Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA Draft guidance https://www.fda.gov/media/87219/download
- Diaz, D. A., Colgan, S. T., Langer, C. S., Bandi, N. T., Likar, M. D., & Van Alstine, L. (2016). Dissolution similarity requirements: how similar or dissimilar are the global regulatory expectations? The AAPS journal, 18, 15-22.
About The Author:
 Bilel Khedir works for Opalia Pharma Recordati Group as a pharmaceutical development project manager and quality assurance specialist. He is also a pharmacist and MSc student in drug development. His expertise includes conducting preformulation studies and formulation of drug products, analytical methods validation, in vitro bioequivalence studies, and writing new drug products’ marketing authorization dossiers (CTD format). In his previous role at Opalia as quality assurance pharmacist, he oversaw cleaning validation, including routine cleaning and disinfection activities, and executed continuous improvement strategies. You can reach him at khedir.b@opaliarecordati.com.
Bilel Khedir works for Opalia Pharma Recordati Group as a pharmaceutical development project manager and quality assurance specialist. He is also a pharmacist and MSc student in drug development. His expertise includes conducting preformulation studies and formulation of drug products, analytical methods validation, in vitro bioequivalence studies, and writing new drug products’ marketing authorization dossiers (CTD format). In his previous role at Opalia as quality assurance pharmacist, he oversaw cleaning validation, including routine cleaning and disinfection activities, and executed continuous improvement strategies. You can reach him at khedir.b@opaliarecordati.com.