
Targeting RNA With Small Molecules
By Brock Schuman, Ph.D., Azor Biotek

RNA drug targets have the potential to revolutionize treatment landscapes for nearly any disease indication. Beyond mRNA's dogmatic role as the intermediate in gene expression, non-coding RNA play essential roles in transcription regulation, maturation, and translation. This article briefly explores the potential of RNA as a clinically validated avenue for drug development, methods for design and discovery, and implications for the future of medicine.
Traditional drug development has focused on targeting proteins. The objective is usually to either enhance or suppress protein activity. Often, protein drug targets are under- or overexpressed — an underlying cause not necessarily addressed by targeting the problematic protein. This is a contributing factor to the marked latency of many therapeutics that indirectly alter target expression levels (e.g., ion channels and neurotransmitter receptors) and why more than half of all protein drug targets are themselves direct or indirect transcriptional regulators (e.g., nuclear receptors, kinases, and G-protein coupled receptors).1 In this same vein, regulatory non-coding RNAs such as microRNA (miR; refer to Figure 1) are excellent targets for potentially profound treatment response, as they in turn regulate expression levels of many functionally related transcripts simultaneously to offer an innately combinational therapeutic avenue. Such polyspecificity can be a mixed blessing, as off-targets may be contraindicated, carcinogenic, or lethal. As such, thorough bioinformatic analysis is recommended when selecting a target, even if that target has demonstrated promise in preclinical studies.
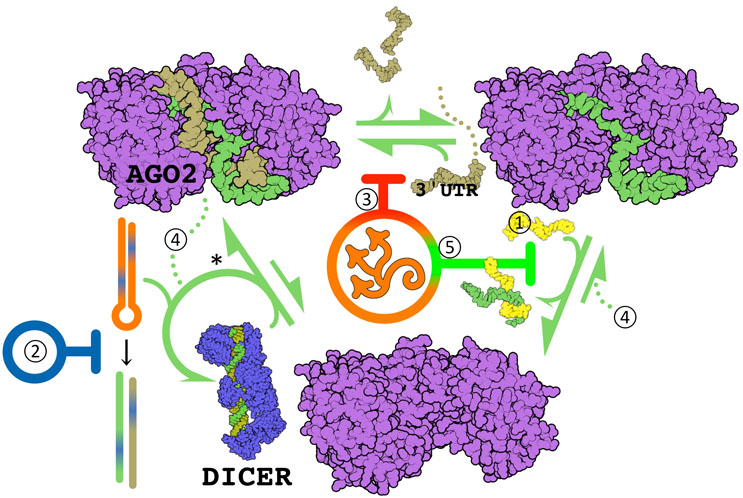
Figure 1: RNA-induced silencing complex targeted therapeutic mechanisms. DICER cleaves pre-miR (hairpin) into the active miR and an imminently cleaved carrier RNA molecule, and facilitates AGO2 docking. miR are guides of the AGO2 endonuclease, where a 5' ≤7 nucleotide seed region hybridizes a target transcript for destruction — usually at mRNA 3' untranslated region (UTR). Hybridization at the miR's 3' end strips it from the active site (as in ①). Overexpressed miR may be downregulated by complementary "antagomiR" oligos (①, e.g., miravirsen2), by inhibition of DICER mediated miR maturation (②, e.g., Targapremir3), or by non-competitive inhibition of hybridization (③, Azor Biotek). Underexpressed miR may be supplemented with oligo "miR mimics" (④, e.g., MRX344), or processivity increased by inhibiting guide discharge (⑤, Azor Biotek). *Each pre-miR species has a major and minor miR product independent of 3' or 5' position — the usual carrier can act as guide. This is a secondary, less efficient mechanism of antagomiR. The figure displays Qutemol26 rendered PDB structures 6mdz27 and 7ele28.
The ephemeral nature of RNA in solution has significantly delayed emergence of new RNA-targeted therapeutics. Furthermore, established early-stage protein-target drug discovery methods are generally incompatible with RNA targets. Computational methods have lagged decades behind equivalent methods for protein targets, high-throughput target engagement studies poorly translate to desirable biological activity, DNA-encoded libraries are particularly unsuitable, and combinatorial chemical libraries are generally inappropriate — every nucleotide exhibits multiple reactive centers in addition to the scissile phosphodiester bonds, far less sheltered by tertiary structure than ordered protein targets. What novel RNA-targeted drugs have emerged are the achievements of target-independent phenotypic screens or functional biochemical screens. As computer-aided drug design techniques advance, we embark upon the era of bespoke RNA-targeted drug design offered by companies like Azor Biotek.
Selectively targeting RNA molecules, it becomes possible to chemically intervene at earlier stages in gene expression to influence protein production and, ultimately, disease progression. The ability to target RNA opens up possibilities to develop therapies to address targets previously considered challenging or undruggable.
Why Small Molecules?
It is comparatively straightforward to target RNA imbalances using RNA molecules as therapeutics — overexpressed targets can be knocked down with complementary oligonucleotides and underexpressed targets can be supplemented with the transcript itself. This simplicity has afforded oligonucleotide therapeutic avenues 97% of allocated development funding compared to small molecules, despite representing <0.5% of the clinically approved market share (2022 market segment valuations as reported in Informa PLC and Beacon Capital). To date, 18 oligonucleotide-therapies have been approved for treatment of various diseases, including elaborate splice-switches and protein-aptamers (reviewed in 5). RNA as both drug and target is appealing given the relative ease of conception, synthesis, and early-stage evaluation — RNA interference regularly demonstrates preclinical promise for treating disease. There are, however, major obstacles to maturing untranslated oligonucleotide therapeutics to clinical use.
1. Bioavailability: Even small (20 nucleotide) oligonucleotides are several orders of magnitude too large and too polar to pass through a cell membrane unaided to reach their target. Furthermore, RNA is readily degraded by RNAses prevalent in most biological compartments. Present advanced delivery vehicles such as lipid nanoparticles may allow access to target sites local to the injection site or the liver, and advancement will continue to expand the accessibility of more physiological compartments. However, administration is limited to injection, severely restricting viability for long-term treatments or personalization of dosage regimens.
2. Competition: Unlike mRNA vaccines that benefit from translational bioamplification, an inhibitory oligo's mechanism of action is limited by the concentration of its overexpressed target — for which it is in direct competition for access to AGO2 along with all other local RNA molecules. RISC assembly is naturally facilitated by DICER to dock freshly matured miR into AGO2 (see Figure 1). Xenobiotic inhibitory oligos often have lower affinity for AGO2 than their complementary RNA targets — meaning the target will induce cleavage of the xenobiotic oligo more efficiently than the xenobiotic will the target. This drives high dosages that exacerbate inherent toxicity.
3. Toxicity: Toxicity might be a beneficial feature, if one were evoking an immune response or targeting tumors. Even then, it is a delicate balance.
Off-target: Especially pertinent for non-coding regulatory RNA therapeutics, serious adverse off-target side effects are a major concern that has led to early termination of numerous clinical trials (e.g., MRX346). This is also a concern that should be evaluated for any small molecule therapeutics, though it is exacerbated by off-target hybridizations and lower potential specificity of oligos.
Formulation: Chemical modifications to mitigate RNA's inherent scissability have been approved for clinical use (e.g., the morpholino Eteplirsen7), but only for severe disease indications as clearance is poor for the large uncleavable molecules, with accumulation and persistence culminating in hepatotoxicity.
Immune: RNA is an immune adjuvant and target. Multiple arms of pattern recognition receptors (e.g., TLRs and RLRs) have evolved specifically to respond to xenobiotic RNA. Immune-mediated adverse reactions have led to the early termination of many oligonucleotide clinical trials, commonly due to pro-inflammatory effects, thrombocytopenia, or thrombosis.8
Biophysical: 1,914 human proteins (8% of the proteome)9 are RNA-binding, homeostatic functioning of which is disrupted by abundant xenobiotic RNA. Furthermore, oligos are inherently sticky molecules: large, labile, polar, massively hydrogen-bonding molecules. They are adhesives, causing hydrogel entanglement upon cellular penetrance by an advanced delivery vehicle (Fig. 3).
4. Specificity: Single nucleotide resolution specificity is rarely achievable with oligonucleotides. Furthermore, off-target effects from biophysical properties and RNA-acting proteins are inescapable.
5. Genericity: Like any biologic, the market share for any oligonucleotide is vulnerable to emulation and market displacement by biosimilars — or preferably by small molecules without oligo-associated caveats.
Small molecules can be designed to mitigate all of these obstacles, increasing precision, bioavailability, and overall safety profiles of treating RNA targets.

Figure 2: RNA-hydrogel entanglement: endosomal escape of RNA from a lipid nanoparticle. A single ~100-nm diameter lipid nanoparticle can house >20 attograms of RNA — only 1/10,000th the mRNA of a typical cell, but concentrated in 0.1% to 2% of the cell's volume. The figure is modified from molecular landscapes of David S. Goodsell 2020 (SARS-CoV-2 mRNA Vaccine; doi: 10.2210/rcsb_pdb/goodsell-gallery-027), and 2020 (SARS-CoV-2 Fusion; doi: 10.2210/rcsb_pdb/goodsell-gallery-026) — illustrations free for use under CC-BY-4.0 license.
RNA-targeted Small Molecules In Clinical Use
We will describe all four classes of clinically approved RNA-targets below and their discovery process. It is noteworthy that all target non-coding regulatory RNA in biological complex with proteins — all 40-odd presently approved molecules have polarity and other physiochemical properties at the cusp of or beyond conventional drug standards.
1. Antibiotics
The prokaryotic 70S ribosome is a marvelous antibiotic drug target: essential and conserved cornerstones to all biological function. Long-established bacterial culture techniques are intrinsically low-cost, high-throughput phenotypic assays suitable for any antibiotic target. Streptomycin was approved for clinical use within a year of discovery in 1944;10 it was the first RNA-acting drug approved by the FDA — though this was not known at the time. Deconvolution of the mechanism of action binding the A-site of the 30S ribosomal subunit to interfere with translational fidelity would not begin in earnest for 20 more years.11 Structural validation and discovery of a secondary P-site blocking mechanism would take over half a century12.
The Nobel Prize meriting crystallization and subsequent structural determinations of the >4.3 megadalton 70S ribosome has elucidated multiple antibiotic-binding sites with diverse mechanisms of action (reviewed in detail elseware13). This structural data has facilitated completely synthetic 70S structure-guided antibiotic design14 and provided the first clinically validated structural data — crucial to train intelligent drug design algorithms.
2. Chemotherapeutics
The 80S ribosomal A-site elongation blocking mechanism as well as antineoplastic properties of omacetaxine mepesuccinate (AKA homoharringtonine) identified from in vitro screens of natural products15 would predate structural elucidation16 and clinical approval for kinase inhibitor resistant leukemia by 35 and 37 years, respectively. It has been suggested that the primary target of omacetaxine mepesuccinate may not be the 80S eukaryotic ribosome as originally described, but the mitochondrial 55S ribosome.17
Translating only 13 proteins, the 55S mammalian mitochondrial ribosomes' three mitochondrial rRNA are encoded by the ribosomal genome. Like 99% of all mitochondrial proteins, ribosomal protein components are encoded by the nuclear genome. Mitochondrial ribosome cross-reactivity has been suggested as a potential chemotherapeutic mechanism for A-site targeting antibiotics, including tetracycline derivatives doxycycline and macrolide azithromycin.18 Despite the challenges associated with accessing the mitochondrial compartment, clinical research in this area is highly active. To date, no antibiotics with 55S cross-reactivity have been approved for reposition as chemotherapeutics.
3. Nonsense Escape
A number of genetic diseases, including Duchenne muscular dystrophy and cystic fibrosis, arise as a consequence of nonsense mutations. These mutations introduce premature stop codons, encoding truncated, non-functional proteins. Phenotypic screens were developed by PTC Therapeutics to identify protein expression rescue in patient-derived bioluminescent reporter cell lines.19 Ataluren was a false positive — a misidentified luciferase stabilizer.20 Clinical viability is marginal (it has conditional approval from the European Medicines Agency but not the FDA); however, its proposed mechanism of action21 merits attention. Akin to streptomycin, ataluren may bind the ribosome's A-site and interfere with translational fidelity — but exclusively at premature stop codons, excluding eukaryotic release factors to allow escape synthesis of the full-length protein.
4. Splice Modification
Spinal muscular atrophy (SMA) is the predominant genetic cause of mortality in infants and toddlers. SMN1 mutations encode low- or non-functional survival motor neuron protein, causing the degeneration of motor neurons, muscle weakness, and, in severe cases — death. SMN1 mutant viability is possible through SMN2 paralog melioration, the copy number of SMN2 being indicative of disease severity.22 However, the SMN protein is still pathologically underexpressed (30%–70%), in part due to SMN2 alternative splicing that excludes exon 7, resulting in a truncated, non-functional protein. Risdiplam, approved by the FDA in 2020, modifies SMN2 splicing to facilitate increased full-length mRNA production, alleviating associated clinical symptoms.
Risdiplam precursors were discovered by high-throughput screening using a human embryonic kidney cell SMN2 bioluminescent reporter line. Hits were further evaluated by full-length SMA transcript expression in SMA patient-derived fibroblast cell cultures, as well as protein expression in motor neuron cultures generated from patient-derived iPSCs (induced pluripotent stem cells).23 While not structurally resolved, the mechanism appears to involve not only SMN2 mRNA binding but direct engagement of U1 spliceosomal RNA and its associated small nuclear ribonucleoprotein.24
Risdiplam is first-in-class not only as a non-ribosomal RNA targeting therapeutic but as the first demonstration of oligonucleotide therapeutic displacement by a small molecule. Nusinersen is an antisense oligonucleotide designed to rectify SMN2 splice variation that was approved for clinical use in 2016.25 Most importantly, risdiplam is associated with longer survival and more motor function gains — but, also, delivery is oral vs. intrathecal, and reported side effects are much less severe.
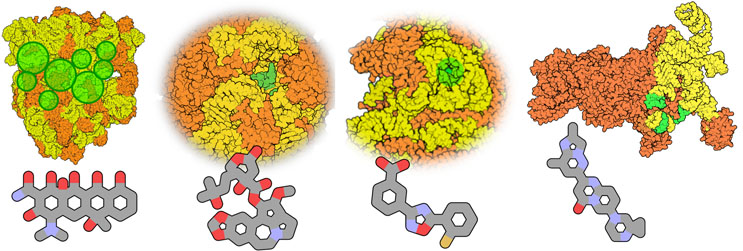
Figure 3: Structures of four clinically approved RNA target complexes in clinical use. Left to right: The prokaryotic 70S ribosome (target of many antibiotics, tetracycline displayed); the mitochondrial 55S ribosome (proposed target of displayed omacetaxine mepesuccinate); A-site of the 80S eukaryotic ribosome (target of omacetaxine mepesuccinate and perhaps displayed Ataluren); and the U1 spliceosome (target of risdiplam). Orange: protein; yellow: RNA; green: (approximate) RNA drug binding sites. The figure displays PDB structures 7s1g,29 5aj3,30 5gak,31 and 6g90,32 rendered with Qutemol.26
Expanding RNA-targeted Therapeutic Scope
The range of potential applications for RNA-targeted therapies is incredibly diverse and continues to expand due to groundbreaking advancements. Through the selective modulation of RNA, we have the ability to intervene at multiple early stages of gene expression. This approach offers highly specific and potentially personalized methods for treating a wide array of diseases. As research progresses and technology advances, the remarkable potential of RNA-based therapies is poised to revolutionize the field of medicine. This paves the way for managing conditions with unmet treatment needs, especially where protein targets of interest are not pharmacologically capable of being targeted. The use of phenotypic screens has been crucial in identifying RNA-targeting drugs currently in clinical use. As the use of rational design methods for developing novel molecules becomes more common, the efficiency and success rate of screening will significantly improve.
References
- A comprehensive map of molecular drug targets. Santos R, et al. Nat Rev Drug Discov. 2017. PMID: 27910877
- Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Lanford RE, et al. Science. 2010. PMID: 19965718
- Small Molecule Inhibition of microRNA-210 Reprograms an Oncogenic Hypoxic Circuit. Costales MG, et al. J Am Chem Soc. 2017. PMID: 28240549
- Chemical modification and design of anti-miRNA oligonucleotides. Lennox KA, Behlke MA. Gene Ther. 2011. PMID: 21753793
- Chemistry, structure and function of approved oligonucleotide therapeutics. Egli M, et al. Nucleic Acids Res. 2023. PMID: 36881759
- Advances in the discovery of microRNA-based anticancer therapeutics: latest tools and developments. To KKW, et al. Expert Opin Drug Discov. 2020. PMID: 31739699
- Eteplirsen: First Global Approval. Syed YY. Drugs. 2016. PMID: 27807823
- RNA Interference-Induced Innate Immunity, Off-Target Effect, or Immune Adjuvant? Meng Z, et al. Front Immunol. 2017. PMID: 28386261
- The human RBPome: from genes and proteins to human disease. Neelamraju Y, Hashemikhabir S, Janga SC. J Proteomics. 2015. PMID: 25982388
- Control of gram-negative bacteria in experimental animals by streptomycin. Jones D, Metzger HJ, Schatz A, Waksman SA. Science. 1944. PMID: 17788929
- Streptomycinoid antibiotics: synergism by puromycin. White JR, White HL. Science. 1964. PMID: 14197563
- Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Carter AP, et al. Nature. 2000. PMID: 11014183
- The bacterial ribosome as a target for antibiotics. Poehlsgaard J, Douthwaite S. Nat Rev Microbiol. 2005. PMID: 16261170
- A synthetic antibiotic class overcoming bacterial multidrug resistance. Mitcheltree MJ, et al. Nature. 2021. PMID: 34707295
- Inhibition of protein synthesis in intact HeLa cells. Tscherne JS, Pestka S. Antimicrob Agents Chemother. 1975. PMID: 1190754
- U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. Gürel G, et al. J Mol Biol. 2009. PMID: 19362093
- Structure-function insights reveal the human ribosome as a cancer target for antibiotics. Myasnikov AG, et al. Nat Commun. 2016. PMID: 27665925
- Treatment of mucosa associated lymphoid tissue lymphoma with a long-term once-weekly regimen of oral azithromycin: Results from the phase II MALT-A trial. Lagler H, et al. Hematol Oncol. 2019. PMID: 30153341
- PTC124 targets genetic disorders caused by nonsense mutations. Welch EM, et al. Nature. 2007. PMID: 17450125
- Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Auld DS, et al. Proc Natl Acad Sci U S A. 2009. PMID: 19208811
- Ataluren binds to multiple protein synthesis apparatus sites and competitively inhibits release factor-dependent termination. Huang S, et al. Nat Commun. 2022. PMID: 35523781
- Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. Crawford TO, et al. PLoS One. 2012. PMID: 22558076
- Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Naryshkin NA, et al. Science. 2014. PMID: 25104390
- SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Palacino J, et al. Nat Chem Biol. 2015. PMID: 26030728
- Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. Haché M, et al. J Child Neurol. 2016. PMID: 26823478
- Ambient occlusion and edge cueing to enhance real time molecular visualization. Tarini M, Cignoni P, Montani C. IEEE Trans Vis Comput Graph. 2006. PMID: 17080857
- Structural Basis for Target-Directed MicroRNA Degradation. Sheu-Gruttadauria J, et al. Mol Cell. 2019. PMID: 31353209
- Structural basis of microRNA processing by Dicer-like 1. Wei X, et al. Nat Plants. 2021. PMID: 34593993
- Structural basis for context-specific inhibition of translation by oxazolidinone antibiotics. Tsai K, et al. Nat Struct Mol Biol. 2022. PMID: 35165456
- Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Greber BJ, et al. Science. 2015. PMID: 25837512
- Structure of the hypusinylated eukaryotic translation factor eIF-5A bound to the ribosome. Schmidt C, et al. Nucleic Acids Res. 2016. PMID: 26715760
- Prespliceosome structure provides insights into spliceosome assembly and regulation. Plaschka C, Lin PC, Charenton C, Nagai K. Nature. 2018. PMID: 29995849
About The Author:
 Brock Schuman, a structural biologist and figure in the field of RNA-targeted therapeutics, obtained his Ph.D. in biochemistry from the University of Victoria, followed by postdoctoral fellowships at the University of Toronto and the United Health Network of research hospitals (now Unity Health). Recognizing the need for advancements in RNA-targeted drug design and development, he founded Azor Biotek to leverage recent progress in molecular dynamics and artificial intelligence to automate the design of bespoke and highly specific RNA-targeting small molecules, and to develop novel high-throughput assays to evaluate their efficacy.
Brock Schuman, a structural biologist and figure in the field of RNA-targeted therapeutics, obtained his Ph.D. in biochemistry from the University of Victoria, followed by postdoctoral fellowships at the University of Toronto and the United Health Network of research hospitals (now Unity Health). Recognizing the need for advancements in RNA-targeted drug design and development, he founded Azor Biotek to leverage recent progress in molecular dynamics and artificial intelligence to automate the design of bespoke and highly specific RNA-targeting small molecules, and to develop novel high-throughput assays to evaluate their efficacy.