
The Discriminative Power Of Dissolution Methods In The US & Europe
By Bilel Khedir, Opalia Pharma Recordati Group

The dissolution methods found in the FDA dissolution database, the U.S. Pharmacopeia (USP) monographs for finished products, or any other compendial monographs for finished products are very useful since they do not require full validation before implementing. Many small details may make a difference while using these methods. This article will discuss a factor that may be game changing for dissolution testing in quality control or research and development of immediate release tablets: the discriminatory power.
The discriminatory power is determined by manufacturing "bad batches," which are batches manufactured with meaningful changes in critical process parameters (CPPs) or critical material attributes (CMAs) that may have an impact on the drug's bioavailability.1 The bad batches may also be generated with quantitative changes in formulation. In order to be discriminative, the method should detect the bad batches when tested.1 Thus, a dissolution method with a good discriminatory power provides a bigger picture regarding the drug product's quality and performance. Not verifying the discriminatory power of a dissolution method, including compendial methods, may have dangerous consequences in quality control and drug development.
Discriminatory Power Of Dissolution Methods In Quality Control
Per the USP, the dissolution test should in most cases be discriminatory for the critical quality attributes of the product.2 The European Medicines Agency (EMA) states in its Reflection paper on the dissolution specification for generic solid oral immediate release products with systemic action that, ideally, all non-bioequivalents should be detected by the in vitro dissolution test results.1 Thus, the dissolution tests in quality control (QC) are done to ensure batch to batch consistency by detecting changes in CPPs and CMAs, and thereby ensuring acceptable in vivo performance.3
If a dissolution method is not discriminatory, meaningful changes in CPPs and CMAs wouldn't be detected and non-bioequivalent batches will pass the test easily.
The big problem here is that many people in the pharma industry believe that the dissolution methods listed in the USP monographs are applicable without verifying their discriminatory power. In reality, these methods need to be tested in terms of their discriminatory power before use. For example, an evaluation of the discriminatory power of the dissolution method used for tablets containing candesartan cilexetil was conducted on generic products in Egypt, and the authors found that the method was not discriminative.4
The American Perspective: U.S. Pharmacopeia and FDA
For generic drug product manufacturers, the use of USP dissolution methods or even methods listed in the FDA dissolution database is not possible without verification of the discriminatory power.5
It is also worth noting that the acceptance criteria set in the USP monographs are not "one size fits all" and must be determined for your specific product considering release profiles during the development of the product. The Q value and the sampling time are selected based on the discriminatory power for critical quality attributes.6
When developing a dissolution method for an immediate release dosage form, USP recommends deriving specifications from dissolution profiles with the following sampling times: 10 minutes, 15 minutes, 30 minutes, 45 minutes, and 60 minutes. Then the Q value is selected at the first time point (not less than 15 minutes) where at least 85% of the labeled amount of the drug product is dissolved.7
The European Perspective: EMA
According to the EMA, setting the Q value and the time range allowing discrimination should be derived from the biobatch dissolution profile. This will allow extrapolation of the results of the bioequivalence studies to the results of the commercial batches.1
The choice of the Q value and the time point is very well explained in the EMA's Reflection paper on the dissolution specification for generic solid oral immediate release products with systemic action.1
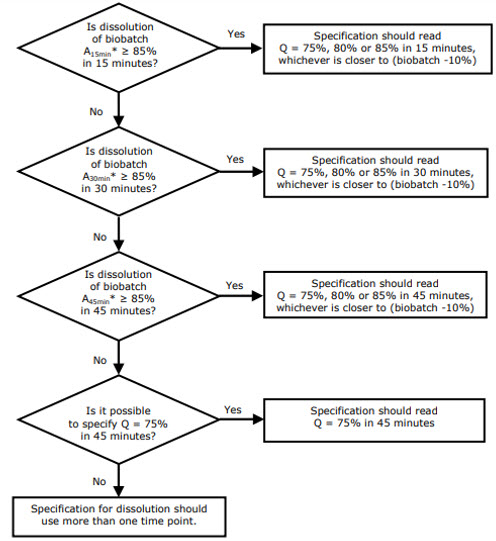
Figure 1: Decision tree for the principles for setting specifications based on the dissolution results of the biobatch.1 "Axmin" stands for "Amount (mean value of 12 units) dissolved in x min".
Discriminatory Power Of Dissolution Methods In Research And Development
In vitro dissolution studies are very important since they are used to select formulations and support bioequivalence studies. Hence, this shows the importance of the discriminatory power of dissolution methods in generic drug product development.
The requirements of the EMA and the FDA regarding the type of dissolution studies for bioequivalence are different for immediate release tablets. The EMA requires dissolution studies in four media: the QC medium and three media with pH1.2, pH4.5, and pH6.8.8 On the other hand, the FDA requires only a dissolution study in the QC medium with discriminatory power studies submitted (even if the the dissolution method used is from USP or the FDA's dissolution database).9
At first glance, the requirements of the FDA may look simple and easy to pass (one medium instead of the four media required by EMA), but in my opinion the American approach is based on good science and, precisely, the discriminatory power concept.
In fact, if the QC dissolution method is discriminative "enough" and your generic drug product shows a similar profile when compared to the brand product, this means that your drug product (generic) is similar in terms of pharmaceutical quality to the brand product, and you can move into a clinical trial with a good level of confidence without conducting studies on the other three media required by the EMA.
The additional media are supposed to mimic the dissolution in vivo, but studies in these media might be challenging (e.g., lack of sink conditions, degradation of the active pharmaceutical ingredient) and they might not reflect what will really happen in vivo for an immediate release tablet.
Discriminatory Power Of A Dissolution Method: Exceptions
The American Perspective : USP and FDA
The USP requires a discriminative dissolution method in most cases.2 The cases where the dissolution method is not required to be discriminative are well-explained in the FDA guideline Dissolution Testing and Acceptance Criteria for Immediate-Release Solid Oral Dosage Form Drug Products Containing High Solubility Drug Substances. In fact, when the drug substance is highly soluble (BCS Class I or III) and formulated in an immediate release form, the developed dissolution method is not required to have discriminative power. As a consequence, you can use the USP dissolution method without verifying the discriminatory power, or you can develop a method as cited in the guideline and submit it without verifying its discriminatory power.10
The European Perspective: EMA
For the EMA, the highly soluble drug substances (BCS Class I or III) make detecting changes in formulation when conducting discriminatory power verification very hard. As a consequence, the dissolution method should be used without further verification or replaced with a disintegration test, as the disintegration test would be more discriminative in this case.1
I think replacing the dissolution test with a disintegration test in this case is very useful and scientifically sound, but it would be very controversial as the European Pharmacopeia in the general monograph of tablets accepts removing disintegration tests when dissolution tests are done for immediate release tablets.
Conclusion
The discriminatory power is a factor often neglected when developing in-house dissolution methods and especially when using compendial methods from finished products monographs. The risks of using a nondiscriminatory method are very significant in QC, as you might be unable to extrapolate bioequivalence results to your commercial batches and, thus, release bad batches. In research and development, using a nondiscriminatory dissolution method might result in selecting a formulation that would fail in clinical trials.
The European and American perspectives regarding dissolution testing in QC and R&D are different, but both emphasize the importance of the discriminatory power of dissolution methods. Since USP and European Pharmacopeia dissolution specifications are not universal, your pharmaceutical company should develop in-house specifications for dissolution and disintegration based on compendial monographs (finished drug products monographs or general monographs) in accordance with studies you've done on your specific product.
References
- Reflection paper on the dissolution specification for generic solid oral immediate release products with systemic action, EMA/CHMP/CVMP/336031 , 2017 https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-dissolution-specification-generic-solid-oral-immediate-release-products-systemic_en.pdf
- USP. Oral Dosage Forms -Performance tests <1711> In : USP–NF. Rockville, MD : United States Pharmacopeia. Accessed February 4, 2023. https://doi.org/10.31003/USPNF_M7083_02_01
- Abend, A., Curran, D., Kuiper, J., Lu, X., Li, H., Hermans, A., ... & Suarez-Sharp, S. (2019). Dissolution testing in drug product development: workshop summary report.
- Amer, A. M., Allam, A. N., & Abdallah, O. Y. (2018). Evaluation of the discriminatory power of USP dissolution method for candesartan cilexetil tablets through testing of marketed products in Egypt. Dissolution Technol, 25(4), 40-46
- Anand, O. M., Yu, L. X., Conner, D. P., & Davit, B. M. (2011). Dissolution testing for generic drugs: an FDA perspective. The AAPS journal, 13, 328-335
- Dissolution Technologies(2017), Question and answers November 2017, accessed January 28th 2023[ https://dissolutiontech.com/issues/201711/201711qa.php]
- USP. The Dissolution Procedure-Development and Validation <1092>. In : USP–NF. Rockville, MD : United States Pharmacopeia. Accessed February 4, 2023. https://doi.org/10.31003/USPNF_M643_05_01
- European Medicine Agency [EMA].(2010) Guideline on the investigation of bioequivalence https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf
- Food and Drug Administration[FDA]. (2021) Bioequivalence Studies With Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA Draft guidance https://www.fda.gov/media/87219/download
- Food and Drug Administration[FDA]. (2018), Dissolution Testing and Acceptance Criteria for Immediate-Release Solid Oral Dosage Form Drug Products Containing High Solubility Drug Substances guidance. https://www.fda.gov/files/drugs/published/Dissolution-Testing-and-Acceptance-Criteria-for-Immediate-Release-Solid-Oral-Dosage-Form-Drug-Products-Containing-High-Solubility-Drug-Substances-Guidance-for-Industry.pdf
About The Author:
 Bilel Khedir works for Opalia Pharma Recordati Group as a pharmaceutical development project manager and quality assurance specialist. He is also a pharmacist and MSc student in drug development. His expertise includes conducting preformulation studies and formulation of drug products, analytical methods validation, in vitro bioequivalence studies, and writing new drug products' marketing authorization dossiers (CTD format). In his previous role at Opalia as quality assurance pharmacist, he oversaw cleaning validation, including routine cleaning and disinfection activities, and executed continuous improvement strategies. You can reach him at khedir.b@opaliarecordati.com.
Bilel Khedir works for Opalia Pharma Recordati Group as a pharmaceutical development project manager and quality assurance specialist. He is also a pharmacist and MSc student in drug development. His expertise includes conducting preformulation studies and formulation of drug products, analytical methods validation, in vitro bioequivalence studies, and writing new drug products' marketing authorization dossiers (CTD format). In his previous role at Opalia as quality assurance pharmacist, he oversaw cleaning validation, including routine cleaning and disinfection activities, and executed continuous improvement strategies. You can reach him at khedir.b@opaliarecordati.com.