
The EU Qualified Person (QP) Demystified: Fool-Proofing Your EU Phase 1 Trial
By Enith Morillo and Azeem Shan

For U.S. sponsors planning to conduct clinical trials in the EU, Qualified Persons (QPs) may at times be perceived as a challenge to overcome, because their role and responsibilities are not fully understood. Conversely, part of the QP’s role is to support clinical trials in the EU by certifying compliant clinical trial materials while protecting public health.
In fact, based on their background, expertise, and legal context, QPs can help expedite the clinical timeline by de-risking unexpected regulatory hurdles that may arise with early-stage novel compounds and complex technology transfers and supply chains, and they can add value when engaged early on.
Therefore, it is imperative to demystify the role of the QP, learn their terminology, and understand what they can and cannot do, in support of Phase 1 clinical trials in the EU. This will ensure gained efficiencies from the get-go, mitigating delays associated with early-stage GMP activities and clinical trial start-up.
This two-part article provides the foundation for U.S. sponsors to understand QPs, their terminology, and the legal context in which they operate to maximize the chances for seamless and effective interactions with them. In Part 2, clear direction is presented to anticipate QP requirements beyond checklists, including drug substance considerations, audits, and shipping excursions handling when limited stability exists.
Meet The QP
European legislation is governed by regulations, directives, and guidelines, with the Qualified Person role being incorporated into European law dating back to 1975 (75/219/EEC). To date, the role of the QP is in several regulatory documents, including current EU directives — 2001/20/EC, 2001/82/EC, and 2001/83/EC, covering human clinical trial materials, veterinary medicinal products, and human medicinal products, respectively.
QPs play an essential role in the EU regulatory landscape and pharmaceutical industry. Although they are best known in the U.S. for performing the QP Declarations and QP Certifications required for conducting clinical trials in the EU, their expertise makes them key players in any organization’s quality unit. Depending on the size and organizational structure of the company, the QP generally serves as a quality director or similar senior position within the quality assurance department, and supports all aspects of the business, including auditing, training, and routine quality functions.
However, unlike in the U.S., where medicinal products are released by the quality unit, in the EU, only a QP (within the quality unit) can certify a batch for use, which is a requirement for release. Through the QP certification, QPs must fulfill their ultimate responsibility: to protect public health.
How To Attain And Maintain QP Status
The prerequisites for becoming a QP include a minimum of extensive scientific education coupled with pharmaceutical GMP experience. QPs are generally either a pharmacist, chemist, or biologist, and in some EU member states, only a pharmacist may become a QP. European legislation is incorporated into national law in each of the 28 member states, such that in the U.K., for example, the role of the QP is defined in U.K. national law, the Human Medicines Regulation. In other EU member states, the same concept applies.
In addition to this prerequisite, EU member states have different QP assessment processes that result in QP eligibility. As an example, in the U.K., one of the most stringent of all, each potential QP must have a “QP sponsor,” an experienced QP who supports the aspiring QP in their pursuit of dedicated theoretical knowledge and practical experience. It is only upon attaining a sufficient scientific baseline that the QP sponsor may endorse the QP candidate to submit his/her application.
Upon submission and review of the application by the respective professional society in the U.K. — i.e., the Royal Pharmaceutical Society, Royal Society of Chemistry, or Royal Society of Biology — QP candidates are invited to attend an interview, commonly known as “QP viva,” to assess the applicant’s knowledge, experience, and soft skills. The assessing panel is made up of three well-rounded, experienced QPs. Successful completion of the interview results in the QP candidate becoming an “eligible QP.”
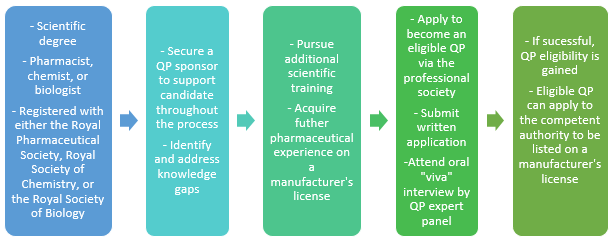
Figure 1: Becoming a QP in the U.K.
For example, the competent authority in the U.K., the Medicines and Healthcare products Regulatory Agency (MHRA), has the ultimate decision whether an eligible QP can be named as a QP on a particular manufacturer’s license. Although QPs may choose to work independently in the industry without being named on a license, only QPs listed on the corresponding manufacturer’s license can certify a batch to a clinical trial, a prerequisite for batch release. It is worth mentioning that a manufacturer’s license is not limited to manufacturers per se, but applies to sites conducting any GMP activities, including testing, packaging, and labeling of medicinal products.
QPs must also abide by a “QP Code of Practice,” ensuring, among other commitments, that the QP only certifies products they have knowledge and expertise in. Where appropriate, QPs may be required to undertake further education and attain adequate experience, prior to certifying batches of a product outside their expertise.
Becoming a QP requires taking a personal vow to adhere to high standards of quality. It requires drive and dedication to attain and commitment to maintain. QPs are required to keep up to date with the evolving nature of the pharmaceutical industry and remain adherent to the QP Code of Practice. If a QP fails to fulfill the obligations entrusted, temporary suspension or administrative or disciplinary procedures may occur.
Learn The QP Terminology
When planning for clinical trials in the EU, there are terms that allow for drawing parallels with those used in U.S. trials and terms that are without equivalents. As an example of parallels, the EU Clinical Trial Application (along with associated supporting documents) is equivalent to the U.S. Investigational New Drug (IND) application; EU competent authorities are the counterparts to the U.S. regulatory authorities, and so on.
However, areas plagued with confusion for U.S. sponsors are the required QP Declaration and QP Certification, as there are no equivalent requirements in the U.S.
The QP Declaration is a document that only a QP can sign and is part of the regulatory submission for a clinical trial in the EU. The objective of this declaration is to attest that non-EU manufacturing site(s) involved in the manufacture of the medicinal product are compliant and up to par with EU GMP standards (Directive 2003/94/EC, 2001/20/EC, EU GMP Part I, etc.). QPs make this declaration only upon thorough and successful verification of this requirement.
On the other hand, QP Certifications are batch-specific documents and are related to EU GMP, the Clinical Trial Application (CTA), and the Product Specification File (PSF). QP certifications involve a thorough and extensive review of batch documentation and clinical documents to “certify” a batch can be released for a clinical trial. It is noteworthy to make the distinction that a QP certification does not equate to batch release, as the latter may be done by non-QP quality personnel. Again, this is dependent on the size and organizational structure of the company.
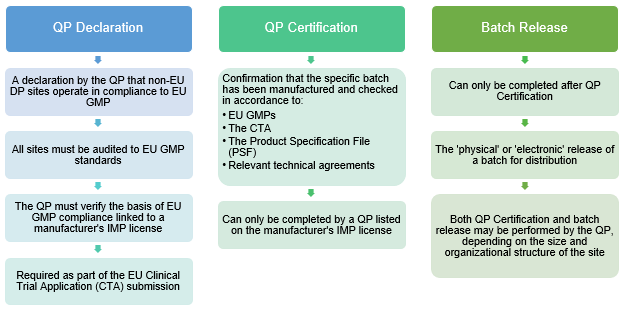
Figure 2: Understanding QP terminology
QP-To-QP Agreements
Depending on the complexity of the supply chain, more than one QP may be involved in the QP certification process, requiring U.S. sponsors to support the negotiation of QP-to-QP agreements. Like Transfer of Regulatory Obligations (TORO) agreements, where delegation of responsibilities is contractually established, QP-to-QP agreements delineate the responsibilities of each QP to ensure there is no ambiguity as to where the responsibility of each QP begins and ends (EU GMP Part I, Chapter 7: Outsourced Activities, EU GMP Annex 16: Certification by a QP and Batch Release, etc.).
As an example, a QP may provide an interim certification for the bulk product, whereby the QP named on the manufacturer’s license for the site where packaging, labeling, and distribution occur performs the final QP certification to the clinical trial. In this case, a QP-to-QP agreement facilitates delineation of responsibilities for each QP and complements the existing technical agreements with the appropriate parties in the supply chain.
Get An Early Start
For U.S. sponsors pursuing clinical trials in the EU, it is imperative that documented responsibilities, unambiguous expectations, and collaborative working relationships are established with QPs early on to facilitate a timely and efficient batch release process. Understanding the role of the EU QP is essential to grasp the extent and responsibility the QP assumes when certifying a batch.
Ultimately, both the U.S. sponsor and the EU QP have a shared responsibility: patient safety.
In Part 2 of this article, the foundation built herein is strengthened with valuable direction for U.S. sponsors on the QP certification process as it relates to drug substance oversight, GMP audit requirements, QP checklists, and shipping excursions handling.
About The Authors:
 Enith Morillo, M.Sc., is the founder and principal consultant of Cadoret Global, a U.S.-based consulting company that specializes in supporting virtual, early stage, and small-sized pharmaceutical companies in taking their investigational drug through development and clinical trials. She has over 12 years of proven, hands-on experience working in the FDA and DEA regulated industry. Morillo specializes in CMC QA oversight of contract manufacturing organizations, batch record review and product disposition, GMP audits, and establishing phase-appropriate quality management systems for emerging pharmaceutical companies. You can reach her at Morillo.Enith@outlook.com or connect with her on LinkedIn.
Enith Morillo, M.Sc., is the founder and principal consultant of Cadoret Global, a U.S.-based consulting company that specializes in supporting virtual, early stage, and small-sized pharmaceutical companies in taking their investigational drug through development and clinical trials. She has over 12 years of proven, hands-on experience working in the FDA and DEA regulated industry. Morillo specializes in CMC QA oversight of contract manufacturing organizations, batch record review and product disposition, GMP audits, and establishing phase-appropriate quality management systems for emerging pharmaceutical companies. You can reach her at Morillo.Enith@outlook.com or connect with her on LinkedIn.
 Azeem Shan, BSc (Hons), MRPharmS, is an experienced EU Qualified Person (QP) based in the UK. A pharmacist by training, he has over a decade of experience in the manufacture, packaging, testing, and distribution of human and veterinary medicines, both as clinical trial materials and commercial products. Shan has first-hand experience with setting up quality management systems, training, performing audits of GDP and GMP facilities, and providing support with remediation issues. His expertise covers EU GMP requirements, including successful MHRA and Veterinary Medicines Directorate inspections, along with site support with FDA pre-approval and GMP inspections. You can reach him at azeem_shan@hotmail.com or connect with him on LinkedIn.
Azeem Shan, BSc (Hons), MRPharmS, is an experienced EU Qualified Person (QP) based in the UK. A pharmacist by training, he has over a decade of experience in the manufacture, packaging, testing, and distribution of human and veterinary medicines, both as clinical trial materials and commercial products. Shan has first-hand experience with setting up quality management systems, training, performing audits of GDP and GMP facilities, and providing support with remediation issues. His expertise covers EU GMP requirements, including successful MHRA and Veterinary Medicines Directorate inspections, along with site support with FDA pre-approval and GMP inspections. You can reach him at azeem_shan@hotmail.com or connect with him on LinkedIn.