
The Importance Of An Analytical Testing Strategy For Combination Products
As combination products evolve and a preference for self-administration systems such as prefilled syringes, auto-injectors, and pen injectors commercialize, there is a need to ensure proper testing of both the primary and secondary components in compliance with regulatory guidelines. For pharmaceutical manufacturers, it is critical to understand the compatibility and performance of the primary packaging system with both the drug product and the delivery systems for successful development of the drug product. Applying a systematic approach and strategy for component and system qualification is important to ensure drug product quality, safety and efficacy. It becomes more challenging to consider performance when a drug product is combined with a delivery system.
The combination product development process to focus on the user-friendliness, or human factors, of self-administration systems, very often involves new and innovative drug products. One crucial aspect of this process focuses on regulatory submission. A foundational point that tends to be overlooked, and required for regulatory submission, is the analytical testing strategy that supports the combination product development process, whether at the stages of concept, feasibility, development, or product release.
Performance Testing
The combination product testing strategy will set the foundation in understanding if requirements will be met and if the combination product will be proven to be safe and effective. The proper development of a combination product testing strategy is not a best practice, but an expected one. It should provide understanding of risks and weaknesses, product design limits, misuse impact as well as environmental impacts on performance. The objective of a proper testing strategy can in one way be achieved by performing the necessary activities to identify a performance issue, if one arises, resolving the issue and proving that the issue has been completely mitigated. A completely thorough assessment of the combination product performance begins with the patient in mind, and closely considers, among other things: (a) regulations, guidances and related requirements; (b) component interactions and mechanical compatibilities; (c) drug compatibility with components; (d) quality and cost targets; and (e) total functional capability with ease of use considerations.
Performance Fundamentals – Historically, the typical containers intended for filling with and storing drug products for later access were vials. The drug content in vials is most often accessed via a syringe. In recent years there have been advancements in drug delivery mechanisms where patients can inject themselves often without the need to access contents in a vial. As personal care has been on the rise in recent years, so has the demand for simpler methods of injection. Syringes, cartridges and devices are now prefilled with drug products. This introduces new challenges for designers and manufacturers of personal injection devices or combination products.
For a given container, such as a prefilled syringe, there are several interfacing components whose fundamental performance must be thoroughly understood when assembled. See example shown in Figure 1, which shows a variety of important performance tests to consider for a finished prefilled syringe. These tests focus on multiple interfaces, some of which can be directly influenced by the drug product, automated assembly, shipping, storage and even final use.
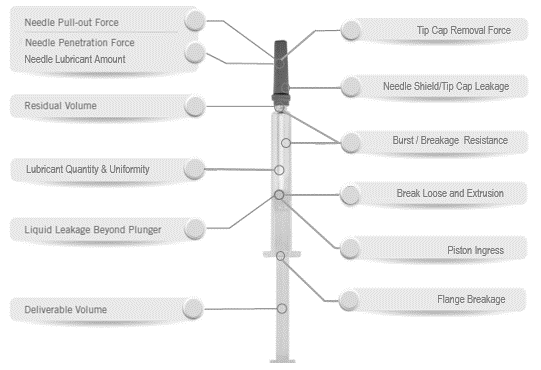
Figure 1: Syringe Testing Considerations
Of the tests to consider, some may be more critical than others depending on several aspects normally captured within a detailed risk assessment. The most critical aspects on which combination product tests are focused are the essential performance requirements (EPRs), which are based on the risk assessment. The EPRs are those requirements that are most important to prove effectiveness from product development through product release, as well as through long term stability testing. Typical EPR examples for a syringe based combination product are: (a) leakage; (b) deliverable volume and (c) break loose and extrusion forces.
Leak Performance – This EPR test is intended to gain understanding and to confirm the potential drug filled syringes leakage related sensitivities as highlighted in Figure 2. This test helps gain leak integrity insight around several areas within a syringe mechanical interfaces (e.g., syringe barrel to plunger, syringe tip cap to needle and syringe tip), as well as higher level interfaces such as in vial adapter systems.
The leak integrity of internal syringe interfaces needs to be thoroughly understood in various conditions in addition to normal use such as environmental conditions like physical shock, vibration, storage temperature, in-flight vacuum and pressure variations. This information is very important since each individual component may be designed very well but may not be perfectly to all mating components specifications and related fit characteristics.
Leaking from a combination product device, or egress, can be a result of either unintended or intended use, but also from filling processing, environmental physical and thermal conditions, drug incompatibility with components and physical incompatibility.
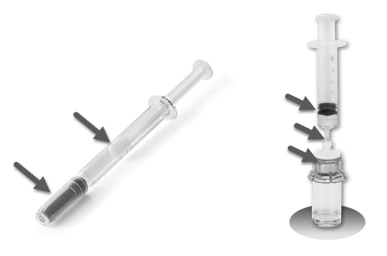
Figure 2: Potential Interfaces Leaks
Deliverable Volume – This EPR test is intended to gain understanding and to confirm if a given syringe, for example, will dose the expected prefilled drug amount. This particular test is greatly influenced by the fill-finish process where the syringe is filled followed by plunger placement. Plunger placement is most typically done with a machine or fixture that creates a vacuum inside of the syringe and releases the vacuum once the plunger is engaged. This action pulls the plunger down to an expected position near the drug products. The filling process consists of a balance of fill volume, desired air gap also known as headspace, and the level of vacuum required to place the plunger in its final desired position.
Deliverable volume errors typically result from errors made in machine vacuum settings, volumetric filling or not properly accommodating or understanding overfill needs. Overfill is the amount of fluid needed to compensate for extra space within the syringe where drug product will remain after dosing, such as the needle and any dead space between the end of the plunger and the bottom inside surface of the syringe. This test is also required to determine if any issues occurred during stability testing and shipping simulation where there could be leaks or premature plunger displacement.
Break Loose and Extrusion – This EPR test is intended to gain understanding of the forces involved with plunger actuation within a prefilled syringe. This test can also be performed to evaluate the uniformity of syringe barrel lubrication, and in this case, it would be performed on a dry syringe. In the case of a prefilled syringe combination product, the break loose and extrusion forces are important to understand as they are directly influenced by drug property interactions with the syringe plunger and syringe barrel inside the lubricated surface. Other factors of significant influence on extrusion or glide forces include the needle inside diameter, drug product viscosity, target injection rate and the syringe inside diameter. As shown in Figure 3, the force curve shows syringe plunger movement starting or initiating force (Fs), the maximum force (Fmax) after initiation, and the mean or average force (F). This force profile is important in that it quantifies force profiles generated due to the fit of the plunger within the syringe barrel while attached to the plunger and associated component interfaces and parameters mentioned previously.
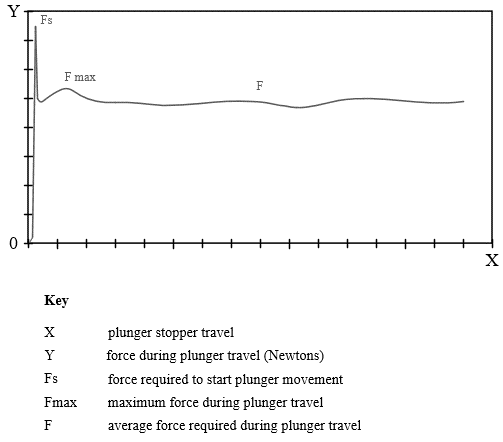
Figure 3: Break Loose and Extrusion Force Profile
Standards and Guidances – When developing a thorough assessment of combination product performance based on perceived risk, where does an organization start? One recommendation is to start with the review and application of the most relevant recognized published guidances and standards. There are several helpful combination product related guidances and standards and are highly recommended to aid in the creation of the testing strategy. Appropriate examples are the ISO 11608 series1 and the 11040 series2.
ISO 116081 is made up of the following parts under the general title needle-based injection systems for medical use according to ISO:
- Part 1: Needle-based injection systems
- Part 2: Needles
- Part 3: Finished containers
- Part 4: Requirements and test methods for electronic and electromechanical pen-injectors
- Part 5: Automated functions
- Part 6: On-body delivery systems
- Part 7: Accessibility for persons with visual impairment
ISO 110402 is made up of the following parts under the general title prefilled syringes according to ISO:
- Part 1: Glass cylinders for dental local anesthetic cartridges
- Part 2: Plungers stoppers for dental local anesthetic cartridges
- Part 3: Seals for dental local anesthetic cartridges
- Part 4: Glass barrels for injectables and sterilized sub-assembled syringes ready for filling
- Part 5: Plunger stoppers for injectables
- Part 6: Plastics barrels for injectables
- Part 7: Packaging systems for sterilized sub-assembled syringes ready for filling
- Part 8: Requirements and test methods for finished pre-filled syringes
All parts of the 116081 and 110402 and series do not directly apply in creating an overall testing strategy for combination products. Several of the ISO standards within both series are insightful and are closely or directly associated with combination product testing aspects, while others within these series focus on components. Below are specific tests, helpful in understanding performance aspects of syringes and cartridges; however, only a number of these tests can be performed at the combination product level. Specific tests are selected based on product knowledge and drug related compatibility.
ISO 11608 Part 31:
- Plunger force
- Leakage
- Dimensions
- Resealability
- Sealability
- Coring
ISO 11040 Part 82:
- Break loose and extrusion forces
- Burst resistance
- Break resistance
- Closure system forces and torques
- Connectivity with fluid path connectors
- Residual volume
- Needle penetration force
- Needle pull-out force
- Sharps injury protection requirements
- Liquid leakage beyond plunger
The ISO 11040-8 and 11608-3 tests above are often performed at the combination product level as a result of a risk assessment and general product knowledge gaps. Most often, there are gaps in understanding drug related interactions with several interfacing components within the combination product. This is due to the variation in drug formulation contents as well as unique component materials not tested with the unique drug product. A fundamental benefit in utilizing standards for the testing strategy is that they often provide insight into not only what to test, but what and how to test. Additionally, most combination product ISO standards are recognized in industry, as well as by regulatory agencies, and are preferred for the testing strategy in comparison to uniquely customized non-standardized testing.
Stability Testing
Combination product stability testing is intended to gain understanding and prove, through data collection and observations, that the quality of the drug and all related interfaces is not negatively impacted by natural environmental influences such as time, temperature and humidity. This is done by establishing re-test periods within a combination product simulated shelf life study based on the expected storage conditions.
For establishing stability testing criteria there are guidances as noted in Table 1. For example, per the World Health Organization ICH Annex 2 WHO Technical Report Series, No. 953, 20093: “At the accelerated storage condition, a minimum of three time points, including the initial and final time points (e.g., 0, 3 and 6 months), from a six-month study is recommended3.” This is a very useful and typical example of stability guidance; however, with proper scientific justification, other approaches to confirm combination product related stability can be utilized based on risk assessment, and product knowledge gaps identified.
Within the same ICH Annex 2 guidance, there are recommendations to test beyond accelerated conditions: “The long-term testing should normally take place over a minimum of 12 months for the number of batches specified in section 2.1.3 at the time of submission and should be continued for a period of time sufficient to cover the proposed re-test period or shelf-life3.”
Table 3: General Case Accelerated Conditions and Durations per ICH Annex 23
|
Study |
Storage Condition |
Minimum Time period Covered by Data at Submission |
|
Long Terma |
25°C ± 2°C/60% RH ± 5% RH or 30°C ± 2°C/65% RH ± 5% RH or 30°C ± 2°C/75% RH ± 5% RH |
12 months or 6 months |
|
Intermediateb |
30°C ± 2°C/65% RH ± 5% RH |
6 months |
|
Accelerated |
40°C ± 2°C/75% RH ± 5% RH |
6 months |
- a - Whether long-term stability studies are performed at 25°C ± 2°C/60% RH ± 5% RH or 30°C ± 2°C/65% RH ± 5% RH or 30°C ± 2°C/75% RH ± 5% RH is determined by the climatic condition under which the API is intended to be stored (see Appendix 1). Testing at a more severe long-term condition can be an alternative to testing condition, i.e., 25°C/60% RH or 30°C/65% RH.
- b - If 30°C ± 2°C/65% RH ± 5% RH or 30°C ± 2°C/75% RH ± 5% RH is the long-term condition there is no intermediate condition.
For combination product stability testing, the frequency of components to be removed from storage simulators (or pulls) occurs at three-month intervals during year one, at six-month intervals at year two, and once annually during year three and beyond. During the pulls, it is generally expected that EPR data is to be collected. It is also expected that general observations should be collected, e.g., changes in drug stability, and usability differences by following instructions for use (IFU), which emulate real-life use scenarios. These extra observations provide more assurances that normal expected-use performance can be achieved throughout the product’s expected shelf-life.
Finally on the subject of stability, per ICH Annex 2: “reduced designs, i.e., matrixing or bracketing, where the testing frequency is reduced, or certain factor combinations are not tested at all, can be applied if justified.3” The primary concern is to ensure EPR can be evaluated especially when a variety of factors are to be considered, e.g., time, temperature and humidity (accelerated and real-time). The ICH Annex 2 guidance is highly recommended to assist in the creation of the stability test program and related strategy creation.
Data Integrity Considerations - What is data integrity and how does it relate to combination products? From the FDA perspective, as noted in the guidance Data Integrity and Compliance with Drug cGMP Questions and Answers (2018), data integrity “refers to the completeness, consistency, and accuracy of data. Complete, consistent, and accurate data should be attributable, legible, contemporaneously recorded, original or a true copy, and accurate (ALCOA).4” When generating combination product testing results and conclusions by following current good manufacturing practices (cGMP), data integrity is very important as these testing results will serve as the primary compilation of proof of viability and will be required for review by the FDA .

Primarily due to recent increases in data audit violations found during cGMP inspections, the FDA has published such guidances. Data integrity is critical for establishing credibility with the FDA regarding combination product safety, effectiveness and overall quality as the data are the main supporting evidence during cGMP inspections. Warning letters from the FDA, consent decrees and import alerts regarding data-related violations are possible and have dire consequences if proper data integrity guidelines are not established and followed. FDA regulations 21 CFR parts 210.15 and 212.26 specify minimum requirements under the Food, Drug and Cosmetic Act for drug related identity, purity, potency and safety, all of which are to be considered in the device. Additional regulations such as 21 CFR part 2117 and part 2126 contain detailed guidances and examples specific to the topic of data integrity. Some examples include the following:
- 21 CFR 212.110(b) - Requiring that data be “stored to prevent deterioration or loss”.6
- 21 CFR 211.100 and 211.160 - Requiring that certain activities be “documented at the time of performance” and that laboratory controls be “scientifically sound”.7
- 21 CFR 211.68 - Requiring that “backup data are exact and complete” and “secure from alteration, inadvertent erasures, or loss” and that “output from the computer… be checked for accuracy”.7
- 21 CFR 211.180 - Requiring that records be retained as “original records,” or “true copies,” or other “accurate reproductions of the original records”.7
- 21 CFR 211.188, 211.194, and 212.60(g) - Requiring “complete information,” “complete data derived from all tests,” “complete record of all data,” and “complete records of all tests performed”.7
The guidances point to data completeness, consistency, and accuracy as being equally as important as the quality, thoroughness, design and robustness of the combination product testing strategy. Data management is critical in having credibility with the FDA for filing and audit purposes. Perhaps even more importantly, scientifically sound data and related documentation ultimately provides a factual and substantive measure of performance to be used for periodic review as needed and future formal audits.
Summary
Combination products by design have become more complex as a result of advancements in drug formulations, container technologies, and related materials – all of which are often independently designed and developed. A variety of components and materials are merged into a final drug product, stored over long periods in various environmental conditions, and then expected to deliver drug therapy safely and effectively. The key goal is to ensure simple, safe and efficacious drug delivery to the patient despite any complexities of producing the quality combination product. Having the proper and well thought out combination product testing strategy rooted in applicable guidances in sync with scientifically sound methods, data capture, and reporting and retainment is no longer a best practice, but an expected one today.
References
- ISO Series 11608 – Package Integrity Evaluation – Sterile Products
- ISO Series 11040 – Pre-filled Syringes
- ICH Annex 2 – Stability testing of active pharmaceutical ingredients and finished pharmaceutical products
- Data Integrity and Compliance with Drug CGMP Questions and Answers Guidance for Industry Guidance for Industry December 2018
- 21 CFR part 210.1 – Status of current good manufacturing practice regulations
- 21 CFR part 212.2 – Applicability of current good manufacturing practice regulations.
21 CFR part 211 – Current good manufacturing practice for finished pharmaceuticals