
The Importance Of Primary And Secondary Drying And Their Impact On Lyophilization
By Sandeep Desai, independent expert

Lyophilization is indispensable for preserving biologics, vaccines, and parenteral formulations. The process converts aqueous solutions into stable solids by removing 95%–99% of the water. It minimizes chemical hydrolysis and enzymatic activity and protects active ingredients from thermal degradation. The process begins with sample freezing, where ice crystals form. This step sets the stage for effective drying. Next, primary drying removes free water through sublimation under low pressure. Temperature and pressure must align precisely to avoid product collapse.
Then, secondary drying removes water that binds to the amorphous matrix. It uses controlled temperature increases to achieve desorption. The goal is to reduce residual moisture to below 1%. Deviations in these steps can cause void formation or semistoppering. Such defects compromise sterility and therapeutic efficacy.1 Regulatory agencies demand strict control of these phases under good manufacturing practices.
Researchers and engineers continuously optimize process parameters using advanced analytical methods to monitor ice crystal morphology, sublimation kinetics, and moisture content. This article analyzes the lyophilization workflow and discusses the mechanistic relation between drying phases and their impact on lyophilization performance. In doing so, it shows the importance of precise process control for product quality.
Complete Lyophilization Process Overview
The lyophilization process involves multiple stages that work together to remove water from frozen solutions. Each stage requires precise control of temperature, vacuum, and duration. Figure 1 shows the basic steps.1
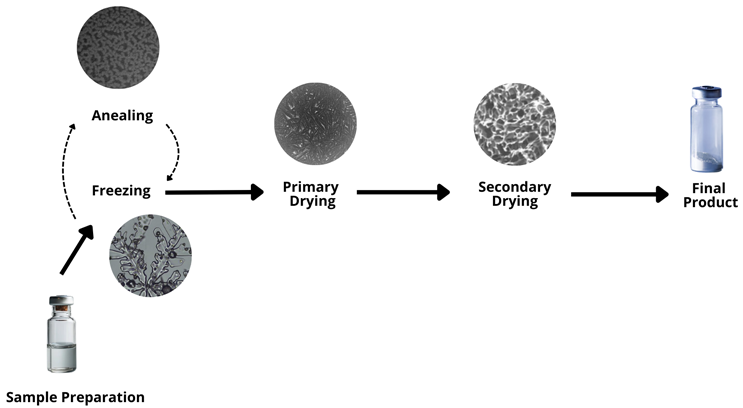
Figure 1: Steps involved in lyophilization from sample preparation to final product.
The figure also shows how the sample changes physically during each stage. In this section of the article, I discuss the core phases of the process and examine the influence of sample preparation, freezing, annealing, primary drying, and secondary drying. I also explore how these steps affect the final product’s quality and stability.
Researchers emphasize that deviations in any phase can induce structural collapse, semistoppering, or void formation. Therefore, operators must optimize process parameters to preserve product integrity and ensure regulatory compliance.2
Sample Preparation
Sample preparation is critical. Scientists first formulate the solution with active ingredients, buffers, and excipients. They choose stabilizers that protect proteins or vaccines from denaturation, and they often include bulking agents to support cake structure. They also add cryoprotectants to minimize ice crystal damage.1,2
These components influence the glass transition temperature and the collapse temperature. The solution must remain below these critical thermal points to avoid structural damage during freezing. Researchers typically sterilize the solution by using filtration. They then dispense it into vials or trays under aseptic conditions.
The container closure system usually includes semistoppers that permit vapor escape while limiting contamination. Operators place the vials on the lyophilizer shelf, ensuring even distribution to promote uniform freezing and sublimation. Any inconsistency in fill volume or vial arrangement can alter heat transfer,3 which leads to uneven drying. Proper sample preparation lays the foundation for a robust lyophilization cycle.4
Freezing
Freezing solidifies the solution into ice crystals and drives the initial phase of water removal. This step must proceed below the glass transition temperature to form a rigid matrix. The process begins with rapid cooling, often to around minus 40 degrees C or lower, depending on the formulation. During this time, ice crystals nucleate and grow. The size of these crystals affects pore structure and subsequent sublimation kinetics.4
Larger ice crystals form when cooling is slower, but extremely slow cooling may encourage solute migration. Rapid cooling can generate smaller crystals, which may improve drying speed but increase surface area for sublimation.1,4
Annealing is an optional procedure that modifies ice crystal morphology. It involves holding the frozen product at a temperature slightly above the initial freezing point but still below the glass transition temperature.1,3 This pause promotes crystal growth and reduces metastable glassy regions. Some formulations also require annealing after the initial freezing. This second annealing step can help reorganize solutes and reduce product heterogeneity. Annealing can improve pore connectivity, leading to more efficient sublimation.3
However, improper annealing can cause partial thawing or collapse. Operators must confirm the safe temperature range for annealing through differential scanning calorimetry or freeze-drying microscopy.5
Primary Drying
Primary drying removes the bulk of the ice via sublimation. Operators apply a high vacuum in the chamber. They also control shelf temperature to drive heat into the product. The sublimation front progresses from the top of the frozen layer toward the vial bottom. The driving force is the vapor pressure difference between the ice interface and the chamber.
Product temperature must stay below the collapse temperature. Exceeding that threshold leads to melt-back or cake shrinkage. The chamber pressure must also remain low enough to facilitate sublimation. Typical pressures range from 50 to 300 mTorr, depending on the product’s sensitivity. The duration of primary drying depends on the vial fill volume, the container geometry, and the thermal properties of the matrix.3
If operators shorten primary drying, residual ice remains. This can lead to incomplete sublimation and potential microbial growth. If they overextend it, product components may degrade due to prolonged exposure to heat. Control systems measure product temperature with thermocouples or wireless sensors. They monitor pressure with Pirani and capacitance manometers.
These measurements guide adjustments to ensure optimal sublimation. Engineers may also use a pressure rise test or tunable diode laser absorption spectroscopy to confirm the endpoint of primary drying.5
Secondary Drying
Secondary drying removes bound or adsorbed water that remains in the dried matrix. This phase typically occurs at higher shelf temperatures than primary drying. Desorption requires additional thermal energy to break water-matrix interactions. Residual moisture levels often drop below 1%.
If operators do not remove enough moisture, the product risks hydrolytic degradation or microbial proliferation. If they remove too much moisture, they may compromise the product’s stability by altering its conformation.6
Proteins can unfold and excipients can degrade. Precise ramp rates help avoid thermal shock. During secondary drying, vacuum levels remain low to enhance mass transfer. The endpoint depends on validated moisture specifications, which are often measured by Karl Fischer titration or near-infrared spectroscopy.7
Careful monitoring is crucial. Any misstep here can diminish the product’s shelf life or therapeutic efficacy.
Final Product
The final product emerges as a porous cake that has a defined structure that allows rapid reconstitution. Operators return the chamber to atmospheric pressure at the end of secondary drying. They then fully stopper the vials to maintain sterility. The residual moisture content, cake appearance, and reconstitution time must meet predefined acceptance criteria.
This final state should show minimal degradation or structural defects. Well-optimized cycles yield products with consistent morphology and stable potency. Any deviation in freezing, primary drying, or secondary drying can undermine these quality attributes. Therefore, scientists validate each step by employing in-process controls and post-process testing. They ensure that the final product remains safe, efficacious, and stable throughout its shelf life.
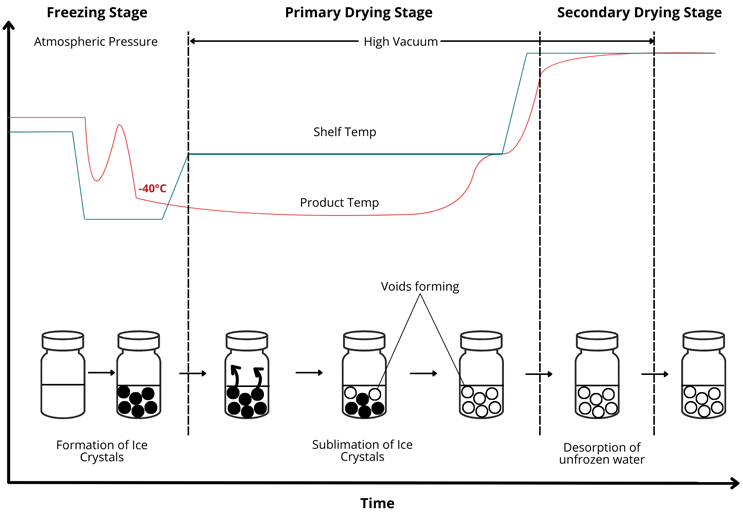
Figure 2: Steps of the lyophilization process and the pharmaceutical freeze-drying equipment refrigeration subsystem
In Figure 2, you can observe the temperature and pressure trends over time. The semistoppering phase allows water vapor to escape while limiting contamination. The void formation risk increases if sublimation or desorption proceeds unevenly.
Proper cycle design reduces this risk. Operators must keep the product temperature below the collapse temperature. They must also manage shelf temperature transitions carefully. This ensures that each step proceeds at an optimal rate. The entire workflow depends on understanding the physicochemical properties of the formulation. It also relies on advanced equipment capable of precise temperature and pressure control.6
Importance Of Primary And Secondary Drying For The Process
Primary and secondary drying are cornerstones of the lyophilization cycle. They dictate product stability, reconstitution properties, and manufacturing efficiency. Their relation determines how well the product maintains its structure and therapeutic efficacy. Operators must balance sublimation in primary drying with desorption in secondary drying. They must also prevent microbial proliferation and degradation.
Researchers state that there is an absolute need for reliable monitoring tools such as manometric temperature measurement (MTM) and tunable diode laser absorption spectroscopy (TDLAS). These tools help identify the end of each drying phase and control critical parameters such as chamber pressure, shelf temperature, and product temperature. 5
Precise adjustments in these stages ensure consistent outcomes and compliance with good manufacturing practices. Table 1, for instance, outlines key parameters and their distinct impacts during primary and secondary drying, but it is important to note that the information presented here is not all-inclusive.4,7
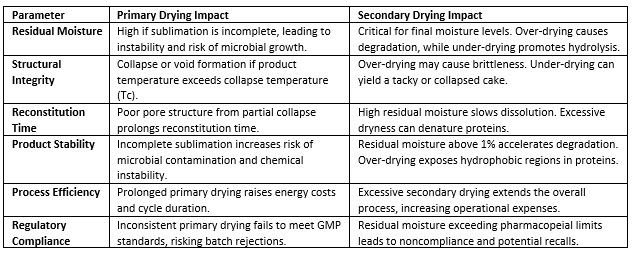
Table 1: Outcomes and impacts of primary and secondary drying
Primary drying sets the stage by removing free ice through sublimation under reduced pressure. Operators control shelf temperature carefully, keeping the product below its collapse temperature to avoid structural deformation. If the product temperature rises above the critical threshold, the matrix may collapse.
This collapse causes a dense or rubbery interior that impedes water vapor flow and prolongs subsequent drying steps. It also distorts the cake, making reconstitution slower and more difficult. Technicians often use TDLAS or MTM to confirm the endpoint of primary drying by measuring the vapor flux or the pressure change.
They adjust shelf temperature to match the sublimation rate. If they shorten primary drying too much, ice remains. That leftover ice may melt during secondary drying, and the melted water rebinds to the product and fosters microbial growth. It also raises the residual moisture content above acceptable limits.1,3,5
Secondary drying focuses on desorbing bound water from the amorphous matrix. Operators raise the shelf temperature above the glass transition temperature but maintain a safe margin to prevent thermal degradation. This step must achieve a delicate balance. If they remove too much water, sensitive proteins may unfold.
Excipients may degrade, leading to potency loss or aggregation. Over-drying also can create a brittle cake structure that fractures easily during transportation. If the operators fail to remove enough water, residual moisture accelerates hydrolysis, oxidation, or even supports bacterial growth.
Researchers often target a final moisture level below 1%. They validate this target with Karl Fischer titration or near-infrared spectroscopy. If the final product exceeds these specifications, it may fail stability tests or become subject to regulatory action.
The structural integrity of the product hinges on both phases. The pore network established in primary drying permits efficient water vapor flow. If voids form unevenly, some regions remain partially wet. Those wet spots cause a non-homogeneous cake with variable moisture content. They also compromise sterility if pockets of water allow bacterial survival. During secondary drying, the matrix may become brittle if the operator prolongs heating or sets an overly high temperature.6,8
Proteins or peptides can unfold when hydrogen bonds break. This phenomenon changes the conformation of macromolecules and reduces therapeutic activity. Thus, each drying phase must remain precisely tuned to maintain the intended cake morphology and preserve bioactivity.8
Reconstitution time also depends on the pore structure and moisture content left after both phases. A well-designed primary drying step creates channels that allow rapid solvent penetration. If the product partially collapses, these channels shrink. That shrinkage increases reconstitution time and complicates administration in clinical settings.
High residual moisture also can slow dissolution, especially if sticky regions form inside the vial. Over-dried cakes, on the other hand, may unfold proteins. This unfolding reduces solubility and leads to turbid reconstituted solutions or aggregates.
From an operational standpoint, both primary and secondary drying affect cycle time and energy consumption. Primary drying is typically the longest step, consuming significant power to maintain vacuum and supply heat. If the operator extends primary drying excessively, operational costs rise. Similarly, secondary drying adds to the total cycle length.
Extended secondary drying also demands more shelf heating and vacuum maintenance. Overly long cycles strain manufacturing schedules and reduce throughput. They also increase the likelihood of thermal degradation. Manufacturers often rely on real-time feedback and modeling to optimize each phase. They adjust shelf temperature in stages to expedite drying without overshooting critical limits.5,7,8
Recommendations
- Optimize Freezing and Annealing: Implement controlled nucleation techniques to ensure uniform ice crystal size. Use annealing to homogenize crystal distribution and minimize heterogeneity in drying rates.
- Monitor Collapse Temperature (Tc): Conduct DSC studies to determine Tc and set shelf temperatures 2 degrees C to 5 degrees C below this threshold during primary drying.
- Real-Time Process Monitoring: Employ MTM or TDLAS to track sublimation rates and endpoint detection. Adjust parameters dynamically to prevent collapse or incomplete drying.
- Validate Residual Moisture Levels: Use Karl Fischer titration to ensure residual moisture is within the target range (0.5%–1%). Conduct stability studies to correlate moisture content with shelf life.
- Prevent Over-Drying: Gradually increase shelf temperatures during secondary drying to avoid protein denaturation. Use BET analysis to determine optimal desorption conditions.
- Ensure Regulatory Compliance: Adhere to pharmacopeial guidelines for residual moisture and sterility. Document all process parameters for audit readiness.
Conclusion
The lyophilization process is a complex relationship between physical and chemical transformations, with primary and secondary drying serving as its backbone. Primary drying essentially removes free water via sublimation, forming a porous matrix, while secondary drying desorbs bound water, ensuring low residual moisture. Deviations in either phase can compromise product quality, stability, and regulatory compliance.
Precise control of temperature, pressure, and duration during these phases is critical. Advanced monitoring tools and optimized protocols mitigate risks such as collapse, void formation, or over-drying. By understanding the mechanistic relationship between primary and secondary drying, manufacturers can enhance lyophilization efficiency, ensure product efficacy, and meet stringent regulatory standards.
This review shows the importance of integrating scientific rigor with operational precision to achieve successful lyophilization outcomes. Future advancements in process analytical technology (PAT) and modeling tools will further refine this critical pharmaceutical process.
References
- Pansare, S. K., & Patel, S. M. (2019). Lyophilization process design and development: a single-step drying approach. Journal of pharmaceutical sciences, 108(4), 1423-1433.
- Jadhav, T. R., & Moon, R. S. (2015). Review on lyophilization technique. World journal of pharmacy and pharmaceutical sciences, 4(5), 1906-1928.
- Jameel, F., Alexeenko, A., Bhambhani, A., Sacha, G., Zhu, T., Tchessalov, S., ... & Jalal, M. (2021). Recommended best practices for lyophilization validation—2021 part I: process design and modeling. AAPS PharmSciTech, 22(7), 221.
- Carullo, A., & Vallan, A. (2012). Measurement uncertainty issues in freeze-drying processes. Measurement, 45(7), 1706-1712.
- Kumar, S., Sanap, S. N., Vasoya, M., Handa, M., Pandey, P., Khopade, A., & Sawant, K. K. (2024). Application of lyophilization in pharmaceutical injectable formulations: An industry and regulatory perspective. Journal of Drug Delivery Science and Technology, 106089.
- Yoon, K., & Narsimhan, V. (2023). Comparison of vial heat transfer coefficients during the primary and secondary drying stages of freeze-drying. International Journal of Pharmaceutics, 635, 122746.
- Jakobsson, C. (2019). Optimization of modified moisture measurement with Karl Fischer to measure moisture content in freeze-dried enzyme beads for improvement of diagnostic kits.
- Ó’Fágáin, C., & Colliton, K. (2023). Storage and Lyophilization of pure proteins. In Protein Chromatography: Methods and Protocols (pp. 421-475). New York, NY: Springer U.S.
About The Author:
Sandeep Desai has 20+ years of experience in pharmaceutical engineering, specializing in HVAC systems, water systems, and manufacturing process equipment. With years of experience in facility design, qualification, and process optimization, he has implemented advanced HVAC solutions to maintain classified environments and ensure regulatory compliance. Adjacent experience lies in designing, installing, and optimizing purified water, WFI, and clean steam systems to meet stringent pharmaceutical standards. He has led modifications and enhancements in filling lines, lyophilizers, autoclaves, and sterilization systems to improve efficiency, reliability, and product integrity. In previous roles, he has worked with OSD and ointment facilities.