
Top 10 Most-Cited MHRA GMP Inspection Deficiencies By Annex/Chapter In 2019
By Barbara Unger, Unger Consulting Inc.
We evaluated the U.K. Medicines and Healthcare products Regulatory Agency’s (MHRA) GMP inspection data for 2019, including trends associated with prior years, in Part 1 of this two-part article. We also addressed the critical and major deficiencies and the annexes and chapters with which they are associated. In this part, we take the 10 chapters and annexes with the highest numbers of deficiencies and do a deeper dive into the specifics of each.
including trends associated with prior years, in Part 1 of this two-part article. We also addressed the critical and major deficiencies and the annexes and chapters with which they are associated. In this part, we take the 10 chapters and annexes with the highest numbers of deficiencies and do a deeper dive into the specifics of each.
In the figures that follow, we identify the paragraphs that were cited most frequently for each of the 10 chapters and annexes identified in Figure 1 of part 1 of this series. For two of these, Chapter 1 and Annex 15, we provide two figures for each because a handful of the paragraphs dominate the deficiencies in those areas. The following figures include all deficiencies, regardless of their classification as critical, major, or other.
In all cases, the table that accompanies each figure lists the paragraph content in the same order as it is found in the figure; they do not follow in numerical sequence.
Chapter 1, Quality System
Let us start with Chapter 1, Quality System, as failure to comply with this chapter is the source of the largest number of total deficiencies, critical deficiencies, and major deficiencies. See also part 1’s Table 2 and Figures 4, 5, and 8. Approximately 25 percent of all deficiencies in 2019 cite Chapter 1.
The data from all deficiencies in Chapter 1 are presented in two figures below because of the disproportionate representation of C1.4 and C1.8 in the total. C1.4 addresses the purpose of the pharmaceutical quality system, and C1.8 addresses, at a very high level, the basic requirements of GMP. Taken together, C1.4 and C1.8 make up 70 percent of the total number of deficiencies that cite Chapter 1 (see the two bars on the left side of Figure 1). Taking a more granular look, the five specific paragraphs representing the bars on the right side in Figure 1 make up 42 percent of all deficiencies that cite Chapter 1. The third through 10th most frequent deficiencies that identify Chapter 1 are presented in Figure 2. The eight paragraphs identified in Figure 2 constitute 27 percent of all deficiencies that cite Chapter 1. Table 1 identifies the text of requirements for the 10 most frequent deficiencies citing Chapter 1.
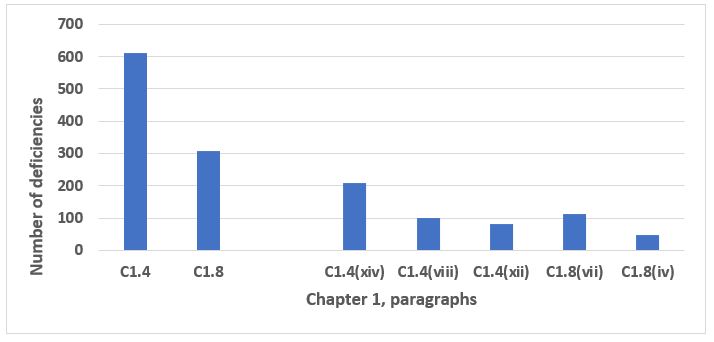
Figure 1: Chapter 1, paragraphs 1.4 and 1.8
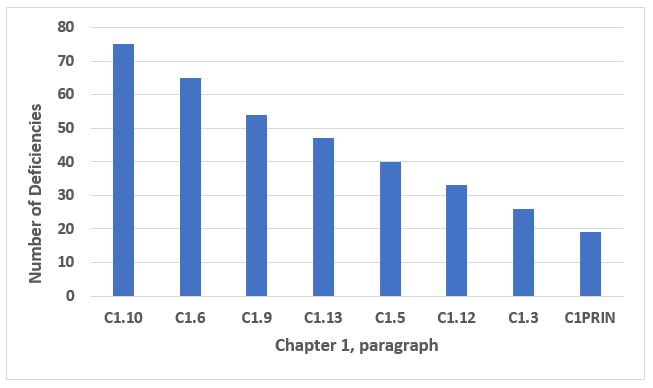
Figure 2: Chapter 1, numbers 3 through 10 in frequency
Table 1: Chapter 1
|
Paragraph |
Short Version of Text |
|
1.4 |
A Pharmaceutical Quality System appropriate for the manufacture of medicinal products should ensure that…(i) through (xvii) |
|
1.8 |
Good Manufacturing Practice is that part of Quality Management which ensures that products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the Marketing Authorisation, Clinical Trial Authorisation or product specification. Good Manufacturing Practice is concerned with both production and quality control. The basic requirements of GMP are that…(i) through (xii) |
|
1.10 |
Regular periodic or rolling quality reviews of all authorised medicinal products, including export only products, should be conducted with the objective of verifying…Such reviews should normally be conducted and documented annually, taking into account previous reviews, and should include at least…(i) through (xii) |
|
1.6 |
There should be periodic management review, with the involvement of senior management, of the operation of the Pharmaceutical Quality System to identify opportunities for continual improvement of products, processes and the system itself. |
|
1.9 |
Quality Control is that part of Good Manufacturing Practice which is concerned with sampling, specifications and testing, and with the organisation, documentation and release procedures which ensure that the necessary and relevant tests are actually carried out and that materials are not released for use, nor products released for sale or 6 supply, until their quality has been judged to be satisfactory. The basic requirements of Quality Control are that…(i) through (viii) |
|
1.13 |
The principles of quality risk management are that…(i) and (ii)… Examples of the processes and applications of quality risk management can be found inter alia in ICH Q9 which is reproduced in Part III of the Guide. |
|
1.5 |
Senior management has the ultimate responsibility to ensure an effective Pharmaceutical Quality System is in place, adequately resourced and that roles, responsibilities, and authorities are defined, communicated and implemented throughout the organisation. Senior management’s leadership and active participation in the Pharmaceutical Quality System is essential… |
|
1.12 |
Quality risk management is a systematic process for the assessment, control, communication and review of risks to the quality of the medicinal product. It can be applied both proactively and retrospectively. |
|
1.3 |
The size and complexity of the company’s activities should be taken into consideration when developing a new Pharmaceutical Quality System or modifying an existing one. The design of the system should incorporate appropriate risk management principles...the effectiveness of the system is normally demonstrated at the site level. |
|
Principle |
The holder of a Manufacturing Authorisation must manufacture medicinal products so as to ensure that they are fit for their intended use, comply with the requirements of the Marketing Authorisation or Clinical Trial Authorisation, as appropriate and do not place patients at risk due to inadequate safety, quality or efficacy…They are described here in order to emphasise their relationships and their fundamental importance to the production and control of medicinal products. |
Chapter 4, Documentation
Running a very distant second place to Chapter 1 is Chapter 4, Documentation, which is cited in approximately 13 percent of all deficiencies in 2019. Figure 3 shows the 10 most frequent citations of Chapter 4. Six of the paragraphs make up almost 75 percent of the citations of Chapter 4, and these can be found in Table 2. Note that 4.8 addresses contemporaneous documentation of GMP records, often recognized in data integrity deficiencies.
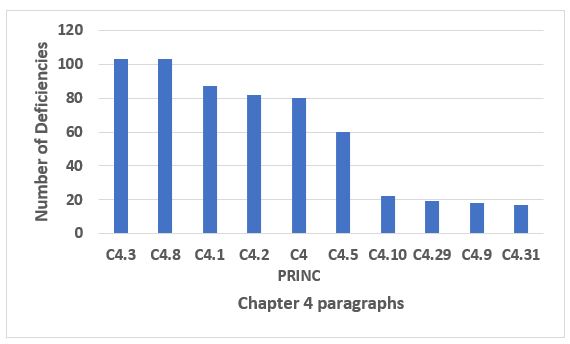
Figure 3: Chapter 4
Table 2: Chapter 4
|
Paragraph |
Short Version of Text |
|
4.3 |
Documents containing instructions should be approved, signed and dated by appropriate and authorised persons. Documents should have unambiguous contents and be uniquely identifiable. The effective date should be defined. |
|
4.8 |
Records should be made or completed at the time each action is taken and in such a way that all significant activities concerning the manufacture of medicinal products are traceable. |
|
4.1 |
All types of document should be defined and adhered to. The requirements apply equally to all forms of document media types. Complex systems need to be understood, well documented, validated, and adequate controls should be in place. Many documents (instructions and/or records) may exist in hybrid forms, i.e. some elements as electronic and others as paper based. Relationships and control measures for master documents, official copies, data handling and records need to be stated for both hybrid and homogenous systems. Appropriate controls for electronic documents such as templates, forms, and master documents should be implemented. Appropriate controls should be in place to ensure the integrity of the record throughout the retention period. |
|
4.2 |
Documents should be designed, prepared, reviewed, and distributed with care. They should comply with the relevant parts of Product Specification Files, Manufacturing and Marketing Authorisation dossiers, as appropriate. The reproduction of working documents from master documents should not allow any error to be introduced through the reproduction process. |
|
C4 Principle |
Good documentation constitutes an essential part of the quality assurance system and is key to operating in compliance with GMP requirements…There are two primary types of documentation used to manage and record GMP compliance: instructions (directions, requirements) and records/reports. Appropriate good documentation practice should be applied with respect to the type of document…Suitable controls should be implemented to ensure the accuracy, integrity, availability and legibility of documents… |
|
4.5 |
Documents within the Quality Management System should be regularly reviewed and kept up-to-date. |
Chapter 5, Production
The top 10 citations of Chapter 5, Production, which make up just under 60 percent of the deficiencies that cite this chapter, are shown in Figure 4. The six most frequent citations in this group constitute 40 percent of the deficiencies for this chapter and are addressed in Table 3. Unlike some of the groups of deficiencies, Chapter 1 and Annex 15, for example, none seem disproportionate in their frequency of citation. Interestingly, the two most frequently cited paragraphs address the use of Quality Risk Management to evaluate and minimize the risks of cross-contamination. In the future, look for this to be a key requirement in the Annex 1 contamination control strategy currently under revision that each firm will need to prepare for.
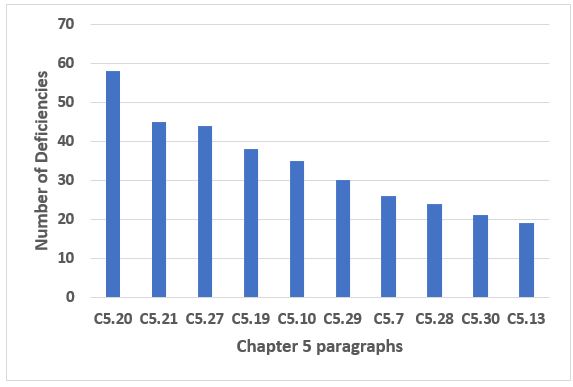
Figure 4: Chapter 5
Table 3: Chapter 5
|
Paragraph |
Short Version of Text |
|
5.20 |
A Quality Risk Management process…should be used to assess and control the cross-contamination risks presented by the products manufactured…The outcome of the Quality Risk Management process should be the basis for determining the necessity for and extent to which premises and equipment should be dedicated to a particular product or product family… |
|
5.21 |
The outcome of the Quality Risk Management process should be the basis for determining the extent of technical and organisational measures required to control risks for cross-contamination. These could include, but are not limited to, the following…Technical Measures (i) through (xiii)…Organizational Measures (i) through (x). |
|
5.27 |
The selection, qualification, approval and maintenance of suppliers of starting materials, together with their purchase and acceptance, should be documented as part of the pharmaceutical quality system. The level of supervision should be proportionate to the risks…Where possible, starting materials should be purchased directly from the manufacturer of the starting material. |
|
5.19 |
Cross-contamination should be prevented by attention to design of the premises and equipment as described in Chapter 3. This should be supported by attention to process design and implementation of any relevant technical or organizational measures, including effective and reproducible cleaning processes to control risk of cross-contamination. |
|
5.10 |
At every stage of processing, products and materials should be protected from microbial and other contamination. |
|
5.29 |
For the approval and maintenance of suppliers of active substances and excipients, the following is required… |
Annex 15, Qualification and Validation
Annex 15, Qualification and Validation, provides an interesting story this year. The four most frequently cited paragraphs constitute 68 percent of all citations from Annex 15. Citations of A15.10, Cleaning Validation, constitute 24 percent of the total. Thus, it seemed appropriate to present those data in more detail. Figure 5 shows the 10 most frequently cited paragraphs in Annex 15, and Table 4 provides the detail for the four most frequent. Figure 6 provides the 10 more frequent citations within A15.10, Cleaning Validation, and Table 5 provides detail for the five areas cited most frequently.
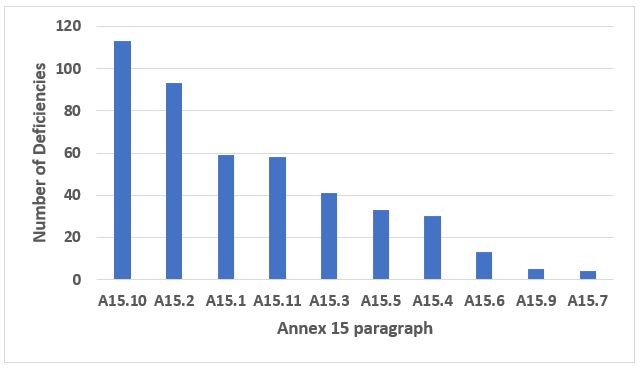
Figure 5: Annex 15
Table 4: Annex 15
|
Paragraph |
Short Version of Text |
|
15.10 |
Cleaning Validation (10.1 through 10.15) |
|
15.2 |
Documentation, Including VMP (2.1 through 2.10) |
|
15.1 |
Organising and Planning for Qualification and Validation (1.1 through 1.8) |
|
15.11 |
Change Control (11.1 through 11.7) |
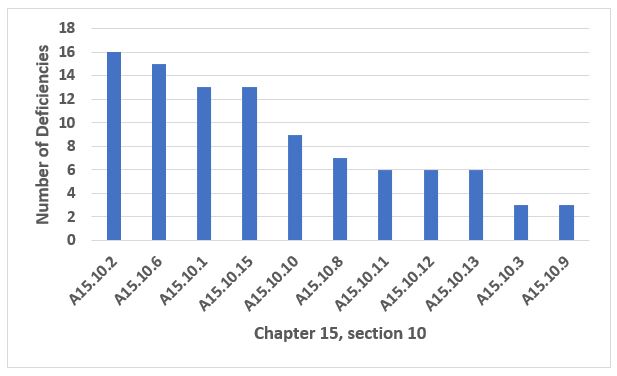
Figure 6: Annex 15.10, Cleaning Validation
Table 5: Annex 15.10
|
Paragraph |
Short Version of Text |
|
15.10.2 |
A visual check for cleanliness is an important part of the acceptance criteria for cleaning validation. It is not generally acceptable for this criterion alone to be used. Repeated cleaning and retesting until acceptable residue results are obtained is not considered an acceptable approach. |
|
15.10.6 |
Limits for the carryover of product residues should be based on a toxicological evaluation. The justification for the selected limits should be documented in a risk assessment which includes all the supporting references…Acceptance criteria should consider the potential cumulative effect of multiple items of equipment in the process equipment train… |
|
15.10.1 |
Cleaning validation should be performed in order to confirm the effectiveness of any cleaning procedure for all product contact equipment. Simulating agents may be used with appropriate scientific justification. Where similar types of equipment are grouped together, a justification of the specific equipment selected for cleaning validation is expected. |
|
15.10.15 |
Changes should be authorised and approved by the responsible persons or relevant functional personnel in accordance with the pharmaceutical quality system. |
|
15.10.10 |
Where a worst case product approach is used as a cleaning validation model, a scientific rationale should be provided for the selection of the worst case product and the impact of new products to the site assessed. Criteria for determining the worst case may include solubility, cleanability, toxicity and potency. |
Chapter 3, Premises and Equipment
Chapter 3, Premises and Equipment, was in fifth place in both 2018 and 2019. While it may seem a bit of a mundane topic, the industry still has plenty of shortcomings in this area. The four most frequent citations from Chapter 3 constitute just under 40 percent of all Chapter 3 deficiencies. The four areas cover equipment (3.34 and 3.41), storage areas (C.19), and general (C.2). Details and text are provided in Table 6.
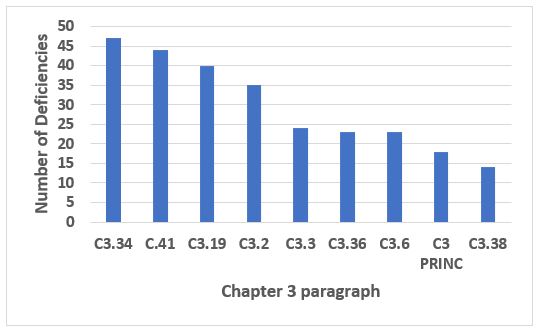
Figure 7: Chapter 3
Table 6: Chapter 3
|
Paragraph |
Short Version of Text |
|
3.34 |
Manufacturing equipment should be designed, located and maintained to suit its intended purpose. |
|
3.41 |
Measuring, weighing, recording and control equipment should be calibrated and checked at defined intervals by appropriate methods. Adequate records of such tests should be maintained. |
|
3.19 |
Storage areas should be designed or adapted to ensure good storage conditions. In particular, they should be clean and dry and maintained within acceptable temperature limits. Where special storage conditions are required (e.g. temperature, humidity) these should be provided, checked and monitored. |
|
3.2 |
Premises should be carefully maintained, ensuring that repair and maintenance operations do not present any hazard to the quality of products. They should be cleaned and, where applicable, disinfected according to detailed written procedures. |
Annex 1, Manufacture of Sterile Medicinal Products
Annex 1, Manufacture of Sterile Medicinal Products, was a close second to Annex 15 in the total number of deficiencies that cite the annexes (see part 1, Figure 3). It was, however, first among the annexes for the number of critical deficiencies in both 2018 and 2019. Annex 1 was a very close second place for the number of major deficiencies, just behind Chapter 1 (see Part 1, Figure 8). The 10 most frequently cited paragraphs from Annex 1 are shown in Figure 8. The four most frequently cited paragraphs constitute approximately 28 percent of all deficiencies citing Annex 1, and the top 10 include 47 percent of all deficiency citations. The text of those four requirements is provided in Table 7.
Looking forward to the revised Annex 1, it will be interesting to see how the deficiencies divide, though I imagine that shortcomings in the company developed contamination control strategy will be near the top of the list.
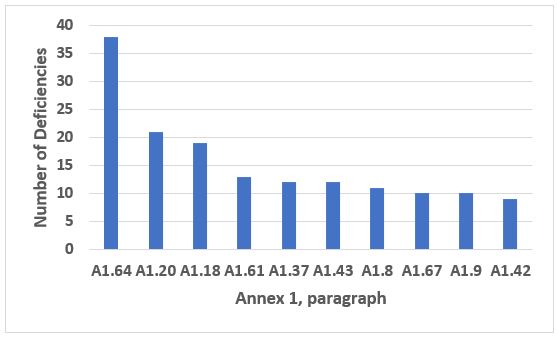
Figure 8: Annex 1
Table 7: Annex 1
|
Paragraph |
Short Version of Text |
|
A1.64 |
Precautions to minimize contamination should be taken during all processing stages including the stages before sterilisation. |
|
A1.20 |
Appropriate alert and action limits should be set for the results of particulate and microbiological monitoring. If these limits are exceeded operating procedures should prescribe corrective action. |
|
A1.18 |
Where aseptic operations are performed monitoring should be frequent using methods such as settle plates, volumetric air and surface sampling (e.g. swabs and contact plates). Sampling methods used in operation should not interfere with zone protection. Results from monitoring should be considered when reviewing batch documentation for finished product release… |
|
A1.61 |
The sanitation of clean areas is particularly important. They should be cleaned thoroughly in accordance with a written programme. Where disinfectants are used, more than one type should be employed. Monitoring should be undertaken regularly in order to detect the development of resistant strains. |
Chapter 8, Complaints and Product Recall
Among the top 10 chapters and annexes, Chapter 8, Complaints and Product Recall, was in seventh place this year. The deficiencies in the top 10 paragraphs shown in Figure 9 constitute 47 percent of all deficiencies citing Chapter 8. Clearly, C8.30 is cited most frequently, approximately the same number of times as the next three in the list, 8.10, 8.13, and 8.15. C8.30 is cited in approximately 18 percent of all deficiencies that cite Chapter 8. Table 8 provides the text of the four most frequent deficiencies that cite Chapter 8.
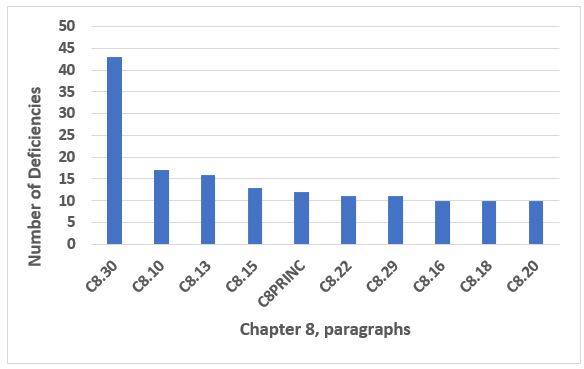
Figure 9: Chapter 8
Table 8: Chapter 8
|
Paragraph |
Short Version |
|
8.30 |
The effectiveness of the arrangements in place for recalls should be periodically evaluated to confirm that they remain robust and fit for use. Such evaluations should extend to both within office-hour situations as well as out-of-office hour situations and, when performing such evaluations, consideration should be given as to whether mock-recall actions should be performed. This evaluation should be documented and justified. |
|
8.10 |
The information reported in relation to possible quality defects should be recorded, including all the original details. The validity and extent of all reported quality defects should be documented and assessed in accordance with Quality Risk Management principles in order to support decisions regarding the degree of investigation and action taken. |
|
8.13 |
The decisions that are made during and following quality defect investigations should reflect the level of risk that is presented by the quality defect as well as the seriousness of any non-compliance with respect to the requirements of the marketing authorisation/product specification file or GMP. Such decisions should be timely to ensure that patient and animal safety is maintained, in a way that is commensurate with the level of risk that is presented by those issues. |
|
8.15 |
Quality defects should be reported in a timely manner by the manufacturer to the marketing authorisation holder/sponsor and all concerned Competent Authorities in cases where the quality defect may result in the recall of the product or in an abnormal restriction in the supply of the product. |
Chapter 6, Quality Control
Chapter 6, Quality Control, is eighth in the top 10 chapters/annexes cited in 2019. The 11 paragraphs identified in Figure 10 (two tied for 10th place) constitute just over 75 percent of all deficiencies that cite this chapter. Paragraphs C6.9 (data trending) and C6.19 (laboratory reagent controls) are the most frequent citations and include 25 percent of all deficiencies that cite Chapter 6.
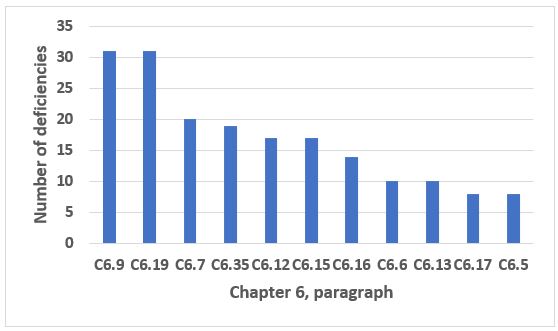
Figure 10: Chapter 6
Table 9: Chapter 6
|
Paragraph |
Short Version |
|
6.9 |
Some kinds of data (e.g., tests results, yields, environmental controls) should be recorded in a manner permitting trend evaluation. Any out of trend or out of specification data should be addressed and subject to investigation. |
|
6.19 |
Special attention should be given to the quality of laboratory reagents, solutions, glassware, reference standards and culture media. They should be prepared and controlled in accordance with written procedures. The level of controls should be commensurate to their use and to the available stability data. |
|
6.7 |
Laboratory documentation should follow the principles given in Chapter 4. An important part of this documentation deals with Quality Control and the following details should be readily 4 available to the Quality Control Department… |
|
6.35 |
Out of specification or significant atypical trends should be investigated. Any confirmed out of specification result, or significant negative trend, affecting product batches released on the market should be reported to the relevant competent authorities. The possible impact on batches on the market should be considered in accordance with Chapter 8 of the GMP Guide and in consultation with the relevant competent authorities. |
Annex 11, Computer Systems
Annex 11, Computer Systems, is frequently cited when the firm does not have adequate controls over electronic systems and data. This annex is frequently associated with the broader data governance and data integrity area. Figure 11 shows the 10 most frequently cited sections from this annex. The section cited most frequently, A11.12, addresses the security of electronic systems and makes up just under 25 percent of the Annex 11 citations. The four most frequent paragraphs, shown in Figure 11, make up slightly over 60 percent of the total and, after security, include validation, data storage, and periodic evaluation of validated systems to ensure they remain in a state of control. The text of these four sections may be found in Table 10.
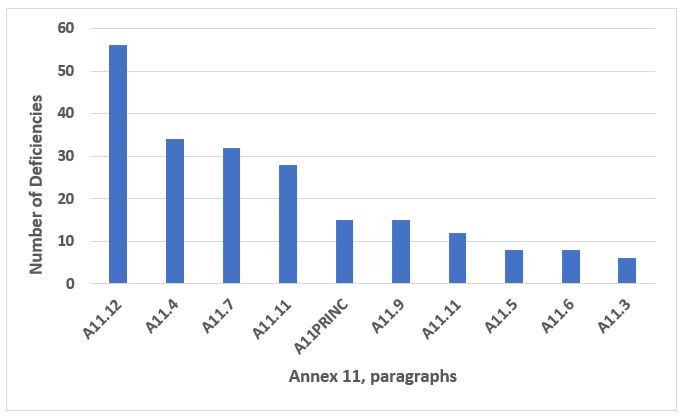
Figure 11: Annex 11
Table 10: Annex 11
|
Paragraph |
Short Version |
|
11.12 |
Security: 12.1 Physical and/or logical controls should be in place to restrict access to computerised system to authorised persons. Suitable methods of preventing unauthorised entry to the system may include the use of keys, pass cards, personal codes with passwords, biometrics, restricted access to computer equipment and data storage areas. 12.2 The extent of security controls depends on the criticality of the computerised system. 12.3 Creation, change, and cancellation of access authorisations should be recorded. 12.4 Management systems for data and for documents should be designed to record the identity of operators entering, changing, confirming or deleting data including date and time. |
|
11.4 |
Validation: 4.1 The validation documentation and reports should cover the relevant steps of the life cycle. … 4.2 Validation documentation should include change control records (if applicable) and reports on any deviations observed during the validation process. 4.3 An up to date listing of all relevant systems and their GMP functionality (inventory) should be available. For critical systems an up to date system description detailing the physical and logical arrangements, data flows and interfaces with other systems or processes, any hardware and software pre-requisites, and security measures should be available. 4.4 User Requirements Specifications should describe the required functions of the computerised system … User requirements should be traceable throughout the life-cycle. 4.5 The regulated user should take all reasonable steps, to ensure that the system has been developed in accordance with an appropriate quality management system. The supplier should be assessed appropriately. 4.6 For the validation of bespoke or customised computerised systems there should be a process in place that ensures the formal assessment and reporting of quality and performance measures for all the life-cycle stages of the system. 4.7 Evidence of appropriate test methods and test scenarios should be demonstrated. Particularly, system (process) parameter limits, data limits and error handling should be considered. Automated testing tools and test environments should have documented assessments for their adequacy. 4.8 If data are transferred to another data format or system, validation should include checks that data are not altered in value and/or meaning during this migration process. |
|
11.7 |
Data Storage: 7.1 Data should be secured by both physical and electronic means against damage. Stored data should be checked for accessibility, readability and accuracy. Access to data should be ensured throughout the retention period. 7.2 Regular back-ups of all relevant data should be done. Integrity and accuracy of backup data and the ability to restore the data should be checked during validation and monitored periodically. |
|
11.11 |
Computerised systems should be periodically evaluated to confirm that they remain in a valid state and are compliant with GMP. Such evaluations should include, where appropriate, the current range of functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security and validation status reports. |
Chapter 7, Outsourced Activities
Last but not least among the top 10 is Chapter 7, Outsourced Activities. With the increasing use of CDMOs and contract laboratories, this remains an important chapter and one where many companies have deficiencies. The three most frequent areas cited constitute just under 50 percent of the total deficiencies in this area, and the text for these is provided in Table 11. Figure 12 shows the frequency of the top 10 Chapter 7 citations, and these make up 90 percent of the citations of Chapter 7.
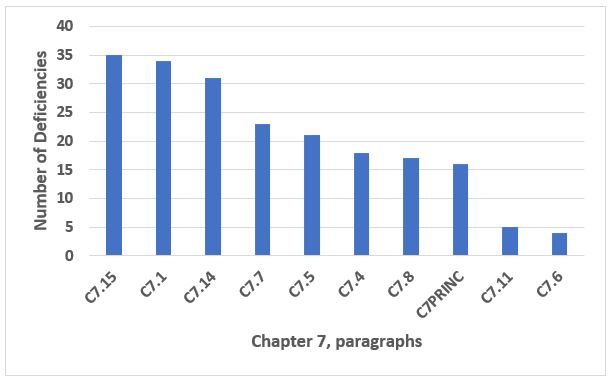
Figure 12: Chapter 7
Table 11: Chapter 7
|
Paragraph |
Short Version |
|
7.15 |
The Contract should describe clearly who undertakes each step of the outsourced activity, e.g. knowledge management, technology transfer, supply chain, subcontracting, quality and purchasing of materials, testing and releasing materials, undertaking production and quality controls (including in-process controls, sampling and analysis). |
|
7.1 |
There should be a written Contract covering the outsourced activities, the products or operations to which they are related, and any technical arrangements made in connection with it. |
|
7.14 |
A Contract should be drawn up between the Contract Giver and the Contract Acceptor which specifies their respective responsibilities and communication processes relating to the outsourced activities. Technical aspects of the Contract should be drawn up by competent persons suitably knowledgeable in related outsourced activities and Good Manufacturing Practice. All arrangements for outsourced activities must be in accordance with regulations in force and the Marketing Authorisation for the product concerned and agreed by both parties. |
Conclusion
This article builds on the conclusions of part 1 that address overall deficiencies, by type and by country over time, considering all chapters and annexes. In part 2, we focus on the 10 most frequently cited chapters/annexes and look at the primary citations within each. In Chapter 1, Quality Systems, deficiencies cluster in two sections, C1.4 and C1.8, addressing fundamental GMP requirements for pharmaceutical firms. We provide additional detail on each of those areas. For Annex 15, Validation and Qualification, much of the focus was on deficiencies in cleaning validation. We take a deep dive into this area in addition to covering the other paragraphs that are cited in Annex 15. Annex 1 is another area where many of the deficiencies cluster in one area regarding failure to take precautions to minimize contamination. And finally, in Annex 11, Computerized Systems, failure to provide adequate system security is the most frequent deficiency.
As we stated at the end of part 1, we hope the MHRA will continue to publish data in the Excel format in the future and expand to include its GCP and GDP inspection findings. It would also be useful for the MHRA to publish the actual text of critical deficiencies from the various areas in future years as it did in the past. But in the absence of that, the publication of these data is valuable and appreciated by the industry.
About The Author:
 Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She was the co-lead of the Rx-360 Data Integrity Working Group. You can contact her at bwunger123@gmail.com.
Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She was the co-lead of the Rx-360 Data Integrity Working Group. You can contact her at bwunger123@gmail.com.