
Transforming Computer System Validation In The Life Sciences Industry
By Partha Anbil and Prashant Deshpande

For decades, traditional CSV has been notoriously burdensome. Teams had to gather exhaustive requirements, run multiple rounds of strict testing (installation, operational, performance qualifications), and produce piles of documentation to prove every step was done right. This manual, document-heavy approach made sense in the past, ensuring critical systems like a manufacturing execution system or quality management software were reliable and compliant. However, it’s become painfully slow and rigid for today’s fast-paced digital world. Here are some examples:
- Long and costly cycles: It’s common for a single large system, such as a new enterprise resource planning software, to require thousands of hours of effort — sometimes 1,500 to 2,000 hours — and six to 12 months to fully validate, costing $1 million to $5 million or more in labor and lost productivity. Paperwork accounts for an estimated 30% of that time.
- High chance of human error: When dozens of spreadsheets, test scripts, and traceability matrices are handled by hand, mistakes inevitably slip in. For example, a simple copy/paste error in a test script or a missing signature can trigger regulatory findings. In fact, incomplete documentation or untracked system changes are frequent culprits behind FDA warning letters for compliance failures.
- Not agile enough: Perhaps most critically, the static “validate once” mindset struggles to keep up with modern IT. Today, many life science firms use cloud-based systems that update every few weeks or integrate streaming data from process analytical technology. Revalidating after each minor update using traditional methods is impractical because teams can’t finish one validation cycle before another update appears. The result is rising risk: companies either fall behind on validation or waste enormous resources to keep up, both unsustainable scenarios in the current fast-paced development landscape.
In short, legacy CSV practices are too slow, too resource-intensive, and too inflexible to meet the needs of an industry where technology is evolving rapidly. The good news? Regulators have noticed these challenges and are modernizing their approach to encourage smarter, leaner validation.
Regulatory Evolution: The 2025–2026 Watershed
Recognizing the limitations of traditional validation approaches and the rapid advancement of digital technologies, regulatory authorities have significantly modernized their guidance. The period between 2025 and 2026 marks a watershed moment for compliance frameworks, shifting the focus from exhaustive documentation to critical thinking and risk-based assurance.
In September, the FDA released its highly anticipated final guidance on Computer Software Assurance (CSA) for Production and Quality System Software, which was subsequently updated in February 2026 to align with the new Quality Management System Regulation (QMSR) and ISO 13485:2016.1 This guidance introduces a modern, less demanding approach to validating software used in manufacturing and quality systems. It explicitly reframes the process around “intended use” and “process risk,” encouraging manufacturers to scale their assurance activities based on whether a software failure could foreseeably compromise patient safety.
Crucially, the 2026 CSA guidance formally recognizes unscripted and exploratory testing as legitimate assurance methods, moving away from the mandate that every test must be rigidly scripted. It also explicitly calls out the application of these principles to automation tools, data analytics, and AI/ML systems used within the quality management system. The guidance endorses leveraging vendor and developer evidence to reduce duplicative validation work and encourages the use of system-generated digital records such as audit trails and logs in place of manual paper artifacts.
The International Society for Pharmaceutical Engineering (ISPE) has also been instrumental in shaping this new era. Building upon the GAMP 5 Guide 2nd Edition released in 2022, the ISPE published its comprehensive GAMP Guide: Artificial Intelligence in July 2025.2 This 290-page framework provides practical, actionable guidance on validating AI- and ML-enabled systems in GxP-regulated environments, addressing everything from model drift and data integrity to algorithmic bias and change control. Furthermore, international harmonization efforts are accelerating. In January 2026, the EMA and the FDA jointly released 10 guiding principles for the responsible use of AI across the medicine development lifecycle, signaling a unified global approach to AI governance in the life sciences.3
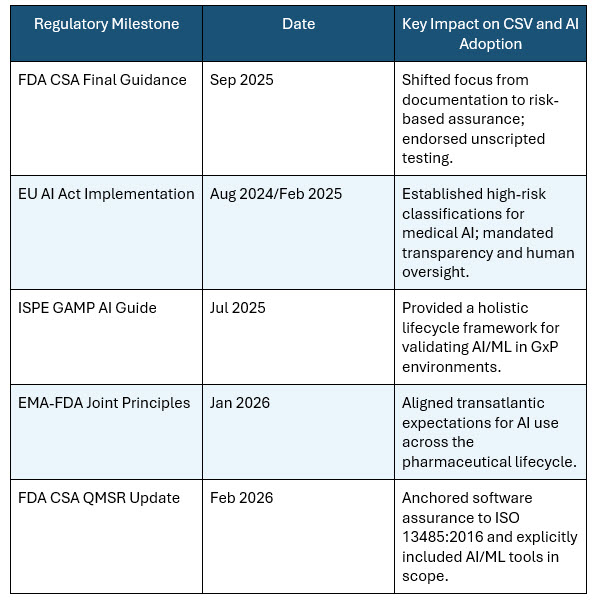
Table 1: Key regulatory milestones shaping AI in CSV (20-2026)
Crucially, these modern approaches explicitly welcome automation and AI. The FDA now allows digital records like system logs or automated test evidence to replace paper printouts and recognizes modern tools, including AI/ML, as valid ways to generate evidence. Industry guidelines like ISPE’s GAMP 5 Guide 2nd Edition and the new GAMP AI Guide provide details on how to manage AI systems within a validated environment, from addressing algorithm bias to controlling continuous learning and changes. In short, the rules have evolved to support innovation as long as companies maintain proper oversight and risk management.
How AI Is Improving CSV With Real-World Examples
With regulatory frameworks now actively supporting innovation, life sciences organizations are rapidly deploying AI to automate and enhance validation workflows, reducing cycle times, and achieving a higher standard of compliance. A 2026 White & Case survey of 200 senior life sciences executives found that 74% consider AI either crucial or particularly important to their business strategy, while 28% plan to invest more than $50 million in AI over the next 12 months.4
Automating documentation and traceability
Documentation has long been the most resource-intensive aspect of CSV. Generative AI and natural language processing (NLP) models are now being utilized to draft validation plans, user requirements specifications (URS), and summary reports using standardized, regulatory-aligned templates. By parsing FDA guidance documents and historical validation artifacts, AI tools can automatically generate comprehensive traceability matrices that link system requirements directly to test cases. The DIA Global Forum reported in November that large software developers are building AI-driven databases of typical user requirements across therapeutic areas, which subject matter experts and quality assurance professionals can customize for their specific needs.5
Generative AI like advanced language models can draft validation documents such as test plans, user requirements, and summary reports, based on templates and historical data. Instead of writing every test case from scratch, an AI tool can propose a comprehensive traceability matrix linking each requirement to tests or suggest a first draft of a validation script by analyzing similar past projects.
For example, one biopharma company used a GPT-based system to generate initial test scripts for 50 analytics dashboards, capturing their filters and data logic. Human reviewers then refined these scripts, focusing only on the tricky, context-specific parts.
The initiative led to a 40% reduction in writing time and more standardized test coverage.6 Across the industry, such AI-driven document drafting is freeing validation experts to spend 35% to 50% less time on rote writing, letting them concentrate on risk assessment and review instead of copy-pasting text.7

Predictive analytics for risk assessment
ML models are transforming risk assessment from a subjective, qualitative exercise into a data-driven, predictive science. By analyzing years of historical validation data, system logs, and audit findings, AI algorithms can predict which software modules or process parameters carry the highest risk of failure.
For example, predictive models utilizing Monte Carlo simulations can identify high-risk components in complex manufacturing execution systems, allowing quality teams to prioritize rigorous scripted testing for those specific areas while employing leaner, unscripted testing for low-risk functions. This targeted approach perfectly aligns with the FDA’s CSA methodology, optimizing resource allocation without compromising safety.
ML models can sift through years of past testing results, incident logs, and manufacturing data to predict where future problems are most likely. This turns risk assessment from a subjective exercise into a data-driven forecast. For instance, a global vaccine manufacturer used ML to analyze historical software glitches across its sites. The model highlighted that one particular laboratory information module, when updated, had caused over 50% of past deviations. Armed with this insight, the validation team focused their efforts on that module’s high-risk features, running extensive tests there while using simpler spot-checks for low-risk areas. This targeted approach — exactly what the FDA’s risk-based CSA encourages — saved time and gave leadership confidence that the biggest risks were under control. By preemptively focusing on known weak points, teams can catch issues before they occur in production, reducing the chance of costly recalls or compliance problems.
Strategies for Moving Forward with AI in CSV
As the life sciences industry transitions from experimental AI pilots to enterprise-scale deployment, organizations must adopt a strategic, structured approach to integrating AI into their compliance and validation workflows. Based on the evolving regulatory landscape and industry best practices, three strategic imperatives emerge.
1. Establish AI Governance and Workforce Enablement
Successful AI adoption requires more than just technology; it requires a cultural shift. Organizations must establish cross-functional AI governance committees comprising IT, quality assurance, regulatory affairs, and legal experts. These committees should be responsible for defining acceptable use cases, assessing vendor AI capabilities, and ensuring alignment with global regulations like the EU AI Act. Furthermore, significant investment must be made in upskilling the existing workforce. Quality and validation professionals need to be trained not as data scientists, but as “AI operators” who understand how to interact with, evaluate, and govern AI systems effectively. As PricewaterhouseCoopers emphasizes, lasting impact requires “smart integration, trusted governance and skilled teams who know how to make it work.”6
2. Adopt a Phased, Risk-Based Implementation Strategy
Organizations should avoid attempting to overhaul their entire quality system simultaneously. A phased approach allows teams to build confidence and refine protocols. Begin by deploying AI in lower-risk, non-GxP areas, such as summarizing internal IT policies or conducting preliminary gap analyses on standard operating procedures (SOPs). Once technology and the internal governance processes are proven, organizations can scale AI applications to high-impact GxP workflows, such as deviation management, CAPA generation, and formal test script automation. McKinsey projects that AI agents could deliver 35% to 45% time savings across clinical development functions, including regulatory affairs and compliance.8
3. Prioritize data architecture and interface integration
The effectiveness of AI in CSV is heavily dependent on the underlying data architecture. Many life sciences companies still rely on legacy systems that lack modern application programming interfaces. Retrofitting AI onto these outdated systems can be prohibitively expensive and technically complex. Strategic IT investments should prioritize modernizing data infrastructure, breaking down data silos, and implementing interface-first architectures that allow AI tools to seamlessly ingest and analyze data across the enterprise.
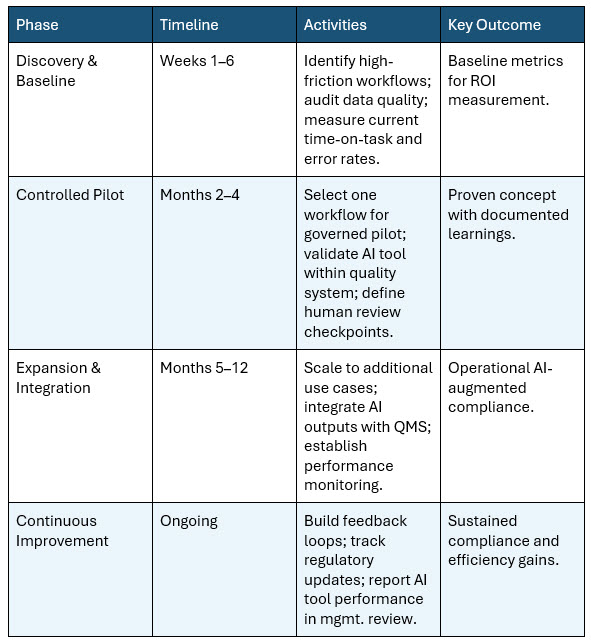
Table 2: Recommended AI-CSV implementation roadmap
Conclusion
The integration of artificial intelligence into computer system validation represents a defining moment for the life sciences industry. By transitioning from manual, documentation-heavy processes to intelligent, risk-based assurance, organizations can dramatically reduce compliance costs, accelerate time-to-market for critical therapies, and enhance overall quality. The regulatory landscape has never been more supportive of this transformation: the FDA’s CSA guidance, the ISPE’s GAMP AI framework, and the EMA-FDA joint principles collectively provide a clear, actionable roadmap for responsible AI adoption.
References:
- PSC Software. (2026). FDA’s Computer Software Assurance 2026: Changes and What to Do Next. https://pscsoftware.com/resource-center/article/fda-computer-software-assurance-2026-changes/
- BioProcess International. (2025). ISPE releases GAMP guide on artificial intelligence. https://www.bioprocessintl.com/regulations/ispe-releases-gamp-guide-on-artificial-intelligence
- IntuitionLabs. (2026). Factors Hindering AI Adoption in Life Sciences: 2023–2026. https://intuitionlabs.ai/articles/ai-adoption-life-sciences-barriers
- White & Case. (2026). New frontiers: How AI is transforming the life sciences industry. https://www.whitecase.com/insight-our-thinking/new-frontiers-how-ai-is-transforming-the-life-sciences-industry/state-of-market
- DIA Global Forum. (2025). The Next AI Revolution: Computer System Validation. https://globalforum.diaglobal.org/issue/november-2025/the-next-ai-revolution-computer-system-validation/
- PwC. (2025). Speed, scale and compliance: How AI is transforming computer system validation (CSV). https://www.pwc.com/us/en/industries/health-industries/library/computer-system-validation.html
- Rook Quality Systems. (2025). FDA Computer Software Assurance 2025 Guidance. https://rookqs.com/blog-rqs/fda-computer-software-assurance-2025-guidance
- Sakara Digital. (2026). AI in Life Sciences Compliance: A Practical Guide for 2026. https://sakaradigital.com/blog/ai-in-life-sciences-compliance-a-practical-guide-for-2026/
- U.S. FDA. (2025). Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-use-artificial-intelligence-support-regulatory-decision-making-drug-and-biological
- IntuitionLabs. (2026). ISPE GAMP AI Guide: Validation Framework for GxP Systems. https://intuitionlabs.ai/articles/ispe-gamp-ai-validation-guide-gxp
Editor's Note: The views expressed in the article are those of the authors and not of the organizations they represent.
About The Authors:
 Partha Anbil is at the intersection of the life sciences industry and management consulting with over 30 years of experience. He is a life sciences industry advisor at MIT, his alma mater. He held senior leadership roles at WNS, IBM, Booz & Company, Symphony, IQVIA, KPMG Consulting, and PWC. He has consulted with and counseled health and life sciences clients on structuring solutions to address strategic, operational, and organizational challenges. He is a diplomat/fellow at MIT CSAIL. He is a healthcare expert member of the World Economic Forum (WEF). He was a member of the IBM Industry Academy, a selective group of professionals inducted into the academy by invitation only, the highest honor at IBM.
Partha Anbil is at the intersection of the life sciences industry and management consulting with over 30 years of experience. He is a life sciences industry advisor at MIT, his alma mater. He held senior leadership roles at WNS, IBM, Booz & Company, Symphony, IQVIA, KPMG Consulting, and PWC. He has consulted with and counseled health and life sciences clients on structuring solutions to address strategic, operational, and organizational challenges. He is a diplomat/fellow at MIT CSAIL. He is a healthcare expert member of the World Economic Forum (WEF). He was a member of the IBM Industry Academy, a selective group of professionals inducted into the academy by invitation only, the highest honor at IBM.
 Prashant Deshpande is an internationally experienced IT executive and consultant to the life sciences industry. He has over 25 years of IT services experience in the digital space and has held senior leadership roles at large-scale IT companies such as Cognizant, Capgemini, L&T Mindtree, and CMC where he led complex delivery transformation programs across industries, including health care and life sciences.
Prashant Deshpande is an internationally experienced IT executive and consultant to the life sciences industry. He has over 25 years of IT services experience in the digital space and has held senior leadership roles at large-scale IT companies such as Cognizant, Capgemini, L&T Mindtree, and CMC where he led complex delivery transformation programs across industries, including health care and life sciences.