
Understanding The FDA's Knowledge-Aided Assessment & Structured Application (KASA) Framework
By Bikash Chatterjee, President and Chief Science Officer, Pharmatech Associates
 In 2004 the FDA put forth its vision for a new approach to ensuring pharmaceutical quality with its landmark final report, Pharmaceutical CGMPs for the 21st Century - A Risk Based Approach.1 The intent behind this initiative was to drive the industry to a quality framework, based upon scientific understanding rather than inspection and testing, but also to enhance and modernize the regulation of pharmaceutical manufacturing and product quality, bringing a 21st-century focus to this critical FDA responsibility. The FDA has never been shy about not only its desire to be more efficient, but also to innovate, as industry has pushed itself to do so.
In 2004 the FDA put forth its vision for a new approach to ensuring pharmaceutical quality with its landmark final report, Pharmaceutical CGMPs for the 21st Century - A Risk Based Approach.1 The intent behind this initiative was to drive the industry to a quality framework, based upon scientific understanding rather than inspection and testing, but also to enhance and modernize the regulation of pharmaceutical manufacturing and product quality, bringing a 21st-century focus to this critical FDA responsibility. The FDA has never been shy about not only its desire to be more efficient, but also to innovate, as industry has pushed itself to do so.
The FDA has continued to evolve its regulatory assessment framework from the summary-based review in the 1990s, through the question-based review2 and risk-based approach3 in the 2000s, to the integrated quality assessment in 2015, and beyond.4 As the complexity of new drug therapies and drug delivery systems has increased, the challenge for the agency to efficiently and consistently oversee pharmaceutical quality has also grown greater. Practically, with the reauthorization of the Prescription Drug User Fee Act (PDUFA VI), Biosimilar User Fee Amendments (BsUFA II), and Generic Drug User Fee Amendments (GDUFA II), the FDA has experienced a large increase in volume of regulatory drug applications along with, in some cases, shorter assessment timelines because of initiatives from the 21st Century Cures Act. To address these challenges and manage the influx of information, the FDA is taking a page from industry’s playbook and is looking to innovate a new system called Knowledge-aided Assessment & Structured Application (KASA).5 The KASA system is designed to:
- Capture and manage information and data during the life cycle of a drug product;
- Establish rules and algorithms for risk assessment, control, and communication;
- Perform computer-aided analyses of applications to compare regulatory standards and quality risks across applications and facilities; and
- Provide a structured assessment that minimizes text-based narratives and summarization of provided information.
More specifically, KASA is intended to capture and manage information about inherent risk and control approaches for product design, manufacturing, and facilities, in a structured format with the intent to facilitate a concise and consistent quality assessment, and largely replace freestyle text. The KASA interface tabulates the following for each critical product quality attribute:
- Inherent risk to quality,
- Control approaches – using a list of generalized structured descriptors related to pharmaceutical design, development, control strategy, and facility implementation,
- A concise summary from the assessor detailing how the generalized approaches are applied in the regulatory application, and
- Links to supporting information from the application.
The FDA’s KASA framework is depicted in Figure 1 below, which highlights the three major components, defined as pillars, that make up the core elements:
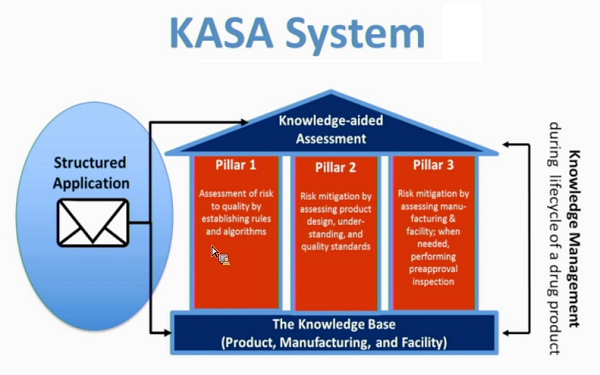
Figure 1: Knowledge-aided Assessment & Structured Application (KASA) system6
KASA Framework
The concept of a house is not new to quality frameworks. Lean manufacturing, with the depiction of a “House of Lean” capturing its core toolkit and principles, is probably the best-known framework. The KASA framework starts with a Knowledge Base intended to encompass the historical information about the drug product and its manufacturing available to the agency.
Pillar 1 – Risk Assessment
KASA uses a predefined ranking table and corresponding algorithms, along with a failure mode, effects, and criticality analysis (FMECA) risk assessment approach to estimate the initial inherent product and manufacturing risks. This is used to objectively and quantitatively assess and rank risks associated with the failure modes of drug product design and manufacturing that have the greatest chance of causing product and manufacturing failure or unexpected harm to the patient. The final risk assessment will be based upon the information provided in the application and may be further informed by the applicant’s risk assessment.
Pillar 2 – Risk Control: Product Design and Quality Standards
Product risk control focuses on the drug substance characteristics and drug product design, understanding, and control. The knowledge captured with such a system enables control of product risk to be compared across applications and facilities as well as evaluate the suitability of the drug sponsor’s proposed specifications.
Pillar 3 – Risk Control: Manufacturing, Facilities, and Inspections
Manufacturing risk control focuses on design and implementation of the manufacturing process and its corresponding control strategy. Facility risk control focuses on a manufacturer’s ability to support the controlled and continued performance of the operations. Evaluation criteria includes the facility’s recent manufacturing history, including applicable Field Alert Reports (FARs), recalls, or regulatory actions.
This remaining risk can be further assessed by performing pre- and post-approval inspections. Under KASA, manufacturing process design and implementation risks will be used to identify the need for pre-approval inspections.
Challenges And Benefits Of KASA
The pursuit of an automated knowledge management system has the potential for great efficiencies, both in the regulatory submission process and in the review process. As with most paradigm-shifting innovations, every solution reveals a new problem to consider. The agency states the current electronic common technical document (eCTD) structure is not fully aligned with the current planned KASA system data structure requirements, raising questions regarding how the data requirements will dovetail with current FDA e-portal requirements and software.
One of the reasons the current framework uses a pdf format is its ability to accommodate multiple data types. If the agency moves to specifying and accepting structured data, is it opening itself up to the same challenges industry faces – managing islands of automation with system specific data lakes? Or perhaps the FDA is seeking to build the new KASA system following an ontological database structure utilizing NoSQL software solutions to accommodate disparate data types and structures? It is a little ironic, but no less admirable, that an organization as IT-challenged as the FDA, which clung for decades to faxing and paper submissions as its foundational method for communication, is equipped to make the leap to the type of infrastructure considerations KASA may present.
However, the FDA may be better positioned than ever in its history to make this leap. The last decade has seen the FDA’s approach to drug regulation evolve, with the agency being forced to participate, not just adjudicate, in the drug development quality discussion. The result has been blockbuster paradigm-breaking drug therapies that do not fall neatly into a regulatory framework, such as antibody-drug conjugates (ADCs), CAR T-cell therapies, next-generation sequencing (NGS), and now human cells, tissues, and cellular and tissue-based products (HCT/Ps), which have pushed the envelope of healthcare worldwide. If a similar approach is taken with KASA, then there is every reason to believe the FDA can be equally successful.
Although KASA is being primarily developed as an assessment tool, it has the potential to greatly streamline the review process and will move the regulatory process toward a structured, defined framework, making it easier for both drug sponsors and evaluators to provide the desired information for review. The structured criteria for evaluation and algorithms could allow assessors to focus on high-risk areas and issues, improving the efficiency of the regulatory evaluation process.
Conclusion
KASA represents an ambitious step forward for the FDA in standardizing and increasing the efficiency of the drug review process. Building the system on a risk framework is consistent with most major regulatory philosophies. For it to be effective, the challenge will be twofold: first, to establish agreed-upon risk evaluation standards that accommodate the myriad technical and quality considerations within a drug submission and that can accommodate the spectrum of current and future drug therapy programs; and, second, to leverage industry’s experience with creating and managing these types of systems.
References:
- Pharmaceutical CGMPs for the 21st Century - A Risk Based Approach - Final Report. September 2004, https://www.fda.gov/media/77391/download
- Yu, L.X., Raw, A., Lionberger, R., Rajagopalan, R., Lee, L.M., Holcombe, F., et al., 2007 US FDA question-based review for generic drugs: a new pharmaceutical quality assessment system. J. Generic Med. 4 (4), 239–246.
- FDA, Guidance for Industry: Q9 Quality Risk Management, 2006.
- Yu, L.X., Woodcock, J., 2015. FDA pharmaceutical quality oversight. Int. J. Pharm. 491, (1-2), 2-7
- Lawrence X. Yu⁎, Andre Raw, Larisa Wu, Christina Capacci-Daniel, Ying Zhang, Susan Rosencrance, FDA’s new pharmaceutical quality initiative: Knowledge-aided assessment & structured applications, https://www.sciencedirect.com/science/article/pii/S2590156719300246?via%3Dihub
- FDA, Slides for the September 20, 2018 Meeting of the Pharmaceutical Science and Clinical Pharmacology (PSCP) Advisory Committee, https://www.fda.gov/advisory-committees/pharmaceutical-science-and-clinical-pharmacology-advisory-committee/slides-september-20-2018-meeting-pharmaceutical-science-and-clinical-pharmacology-pscp-advisory
About The Author:
 Bikash Chatterjee is president and chief science officer for Pharmatech Associates. He has over 30 years’ experience in the design and development of pharmaceutical, biotech, medical device, and IVD products. His work has guided the successful approval and commercialization of over a dozen new products in the U.S. and Europe. Chatterjee is a member of the USP National Advisory Board and is the past-chairman of the Golden Gate Chapter of the American Society of Quality. He is the author of Applying Lean Six Sigma in the Pharmaceutical Industry and is a keynote speaker at international conferences. Chatterjee holds a B.A. in biochemistry and a B.S. in chemical engineering from the University of California at San Diego.
Bikash Chatterjee is president and chief science officer for Pharmatech Associates. He has over 30 years’ experience in the design and development of pharmaceutical, biotech, medical device, and IVD products. His work has guided the successful approval and commercialization of over a dozen new products in the U.S. and Europe. Chatterjee is a member of the USP National Advisory Board and is the past-chairman of the Golden Gate Chapter of the American Society of Quality. He is the author of Applying Lean Six Sigma in the Pharmaceutical Industry and is a keynote speaker at international conferences. Chatterjee holds a B.A. in biochemistry and a B.S. in chemical engineering from the University of California at San Diego.