
Update On Regulatory Strategies To Mitigate Mutagenic Impurities In Prescription Drugs
By Prasanth Venugopalan, Ajay Pazhayattil, and Marzena Ingram

Addressing small molecule woes with nitrosamine and similar impurities is becoming patient critical. The FDA’s MedWatch1 reporting program publishes safety alerts for prescription drug products. In 2019, the N-nitrosodimethylamine (NDMA), N-nitrosodiethylamine (NDEA), and N-nitroso N-methyl 4-amino butyric acid (NMBA) related impurity recall notifications accounted for almost 67% of the total impurity related alerts, the largest category that year. The regulatory scenario and risk mitigation activities associated with implementing control strategies have been evolving ever since. The recalls have been steadily declining (35% for 2020 and 23% for 2021 to date), displaying the effectiveness of the actions initiated. The industry and regulators continue to collaborate2 to maintain drug safety and ensure availability for patients. Alignment on a three-step assessment and impurity control process (Step 1 - assessing the risk, Step 2 - conducting confirmatory testing, and Step 3 - reporting control strategy changes implemented) has greatly helped in streamlining the response.
European Medicines Agency Offers Some Clarity
Recent questions and answers from the European Medicines Agency (EMA)3 provide clarity for the pharmaceutical industry. EMA included products that were approved after Sept.26, 2019, where risk evaluation was not performed as part of the marketing authorization approval process. The risk assessment requirement now applies to all products, irrespective of their marketing status or active supply status, even if Step 2 is not feasible. Performing Step 2 confirmatory testing will be required for those products at the time when batches are manufactured. Submission in this scenario includes a commitment letter and the outcome once the Step 2 testing is completed. Further, the changes made as part of Step 3 need to be submitted prior to relaunching the activated products. The Q&A further clarifies that risk evaluation is required during line extension or variation submissions for the identified products.
The limit for N-nitrosomorpholine (NMOR) has now been established at 127 ng/day. The limit for N-nitroso-N-methyl-4-aminobutyric acid (NMBA) is derived using structure-activity-relationship (SAR)/read-across approach using the TD50 of NDMA as point of departure, while limits for N-nitrosoethylisopropylamine (EIPNA), N-nitrosodiisopropylamine (DIPNA), 1-methyl-4-nitrosopiperazine (MeNP), and N-nitroso-di-n-butylamine (NDBA) are derived using SAR/read-across approach using the TD50 of NDEA as point of departure.
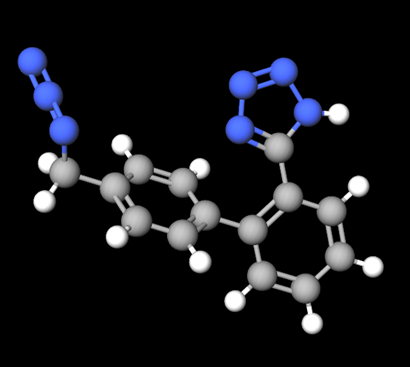
5-(4’-(azidomethyl)-[1, 1’-biphenyl]-2-yl)-1H-tetrazole
Additional Impurities Emerge
Additional mutagenic impurities are emerging since identification of nitrosamine impurities. In 2020, the presence of 5-(4’-(azidomethyl)-[1, 1’-biphenyl]-2-yl)-1H-tetrazole (AZBT) was found in batches of active substance of irbesartan. Detection of the azido impurity resulted in withdrawal4, 5 of irbesartan, losartan, and valsartan batches from different markets across the world. AZBT impurity may occur during synthesis of certain APIs when using common intermediates. AZBT is structurally different from nitrosamine, where the starting material combines with a specific tetrazole ring of active substances. Similar to nitrosamine impurity risk management, testing for this specific impurity in products has been initiated and new validated methods are available to verify the presence of AZBT in products.
Regulatory controls and updates are expected as the industry and regulators become aware of other unknown impurities in well-established products in the market. These occurrences and the potential for revenue loss have already triggered higher focus on process controls and risk management strategies. The level of scrutiny on new product approvals will be influenced by these developments, encouraging the innovators to focus more on their process controls and assess the potential impurities generated during manufacturing. For those products already in the market, the newly learned risk of unknown or unidentified impurities could create global drug shortages while the manufacturers try to optimize their synthesis routes and manufacturing processes.
Challenges Ahead For Manufacturers
Analytical development and testing are other ongoing challenges for manufacturers as the instrumentation, development, validation, and testing costs are swelling. The analytical execution delays will add to the current supply chain slowdown that has emerged from the pandemic. The scenario affects both existing products and small molecule product launch pipelines. The impurity levels that are discussed are in ultra-trace amounts that are not easily detectable and call for complex instrumentation. For nitrosamines, the FDA, Health Canada, and the EMA recommend the use of GC-MS, LC-MS/MS, and GC-MS/MS, and a combination of procedures6, 7, 8, 9 to ensure coverage of a wide spectrum of impurities. Those impurities that were not identified during the development and early-stage stability studies could elicit requirements for higher sensitivity of the analytical methods used. Regulatory agencies across the globe may soon demand more stringent limits for impurities during analytical testing and process controls to remove the impurities generated during the process.
An FDA & Joint Industry Meeting10 was held on May 4, 2021, to review concerns on implementing the FDA’s Guidance for Industry: Control of Nitrosamine Impurities in Human Drugs. The regulator indicated its willingness to revisit the timeline expectations if the reasons are justifiable. The FDA, however, was not willing to extend the risk assessment deadline to Sept. 1, 2021 based on the industry’s request at the time. A similar approach can therefore be expected on risk assessments for any future scenarios. The FDA also indicated its willingness to work with other regulators toward the alignment of expectations, which will be a relief for the industry.
The European Federation of Pharmaceutical Industries and Associations (EFPIA) workflow11 considers the risks from packaging material such as blister foils with lacquer as low risk for nitrosamine formation at this time. However, based on the formulation characteristics and the packaging processes that allow only low volatilization opportunity for NDMA and NDEA, a theoretical risk may arise. Beyond small molecules, biologics such as synthetic peptides may also use reagents that may be of concern. Primary packaging materials for biologics may also contain material that could leach into the product, potentially further expanding the scope into all product categories. The impurity management strategy may extend to manufacturing and testing of key raw materials, intermediates (building blocks), reagents, and starting materials beyond drug substances or drug products.
Conclusion
One of the biggest challenges with nitrosamines is their ease of formation and that various factors in the supply chain can contribute to their formation12. This means that while supply chain controls and patient counseling are essential in avoiding the risks of impurities in drug products, the storage and handling of drugs is critical in preventing impurity formation. To weather the emerging landscape, pharmaceutical manufacturers need specialized focus teams to swiftly determine potential risks and put remedial measures in place. Those actions must be in line with the regulatory accepted assessment and impurity control three-step process.
References
- MedWatch: The FDA Safety Information and Adverse Event Reporting Program:https://www.fda.gov/safety/medwatch-fda-safety-information-and-adverse-event-reporting-program
- Meeting Report: N-Nitrosamine Impurity Control Strategies in the Pharmaceutical and Biotechnology Industries:https://link.springer.com/epdf/10.1208/s12248-021-00618-5
- Questions and answers for marketing authorisation holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products, 29 June 2021 EMA/409815/2020 Rev.4.
- Health Canada advisory; Identification number # RA75715, 15 June 2021.https://healthycanadians.gc.ca/recall-alert-rappel-avis/hc-sc/2021/75715a-eng.php
- Swissmedic; Monitoring of sartan medicines stepped up: traces of a new foreign substance detected, 01 July 2021. https://www.swissmedic.ch/swissmedic/en/home/news/mitteilungen/intensivierte-ueberwachung-sartan-arzneimittel.html
- Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS) Method for the Determination of NDMA in Metformin Drug Substance and Drug Product. www.fda.gov/media/134914/download
- Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Method for the Determination of NDMA in Ranitidine Drug Substance and Solid Dosage Drug Product. www.fda.gov/media/131868/download
- Procedure under Article 5(3) of Regulation EC (No) 726/2004; Nitrosamine impurities in human medicinal products,25 June 2020, EMA/369136/2020
- Health Canada, Nitrosamine impurities in medications: test methods.https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities/test-methods.html
- Memorandum of Meeting, 4 May 2021; FDA & Joint Industry Meeting, Docket FDA-2020-D-1530.https://www.regulations.gov/document/FDA-2020-D-1530-0021
- Workflows for quality risk management of nitrosamine risks in medicines, December, 2020:https://www.efpia.eu/about-medicines/development-of-medicines/regulations-safety-supply/regulatory-affairs/
- FDA Guidance for Industry- Control of Nitrosamine Impurities in Human Drugs, Rev #1, February 2021.
About The Authors:
 Prasanth Venugopalan is manager at Rubicon Research, Canada, and is an industrial pharmacist with an analytical mindset. He holds a master’s degree in pharmacy and has worked with CDMOs and the generic industry in different capacities. He has extensive experience in quality risk management, validation, technology transfer of APIs, and solid dose and nasal formulations.
Prasanth Venugopalan is manager at Rubicon Research, Canada, and is an industrial pharmacist with an analytical mindset. He holds a master’s degree in pharmacy and has worked with CDMOs and the generic industry in different capacities. He has extensive experience in quality risk management, validation, technology transfer of APIs, and solid dose and nasal formulations.
 Ajay Pazhayattil is a pharmaceutical management consultant leading technical operations, quality assurance, and regulatory compliance risk mitigation/remediation projects. He has conceived and implemented novel methodologies applying sound pharmaceutical science principles. He is an industrial pharmacist with experience in solid dose, liquid, and parenteral dosage forms.
Ajay Pazhayattil is a pharmaceutical management consultant leading technical operations, quality assurance, and regulatory compliance risk mitigation/remediation projects. He has conceived and implemented novel methodologies applying sound pharmaceutical science principles. He is an industrial pharmacist with experience in solid dose, liquid, and parenteral dosage forms.
 Marzena Ingram is a senior pharmaceutical consultant at Validant with extensive quality and technical operations experience in solid dose and active pharmaceutical ingredients manufacturing operations. She is currently responsible for delivering compliant solutions for clients. Ingram has developed specialized teams and has spearheaded the implementation of programs to meet global regulatory requirements.
Marzena Ingram is a senior pharmaceutical consultant at Validant with extensive quality and technical operations experience in solid dose and active pharmaceutical ingredients manufacturing operations. She is currently responsible for delivering compliant solutions for clients. Ingram has developed specialized teams and has spearheaded the implementation of programs to meet global regulatory requirements.