
Using Risk-Based Thinking To Manage Nonconformances And Deviations
By Mark Durivage, Quality Systems Compliance LLC
Probably the most significant concern for anyone responsible for implementing, deploying, and maintaining a quality  management system (QMS) is the integration of risk-based thinking. While the concepts of risk-based thinking and management are not new, previous practice was more reactionary, primarily focusing on detection after the fact, root cause analysis, corrective actions, and preventing recurrence of the failure. Contemporary thinking places the emphasis on considering risks up front (prevention) and having a solid approach to address risk in planning, managing, and driving actions.
management system (QMS) is the integration of risk-based thinking. While the concepts of risk-based thinking and management are not new, previous practice was more reactionary, primarily focusing on detection after the fact, root cause analysis, corrective actions, and preventing recurrence of the failure. Contemporary thinking places the emphasis on considering risks up front (prevention) and having a solid approach to address risk in planning, managing, and driving actions.
This article presents the requirements regarding nonconformances and deviations, and then introduces some tools to incorporate and integrate risk management techniques within the QMS, specifically applied to nonconformance and deviation management.
Requirements And Background
There several International Organization for Standardization (ISO) standards, Food and Drug Administration (FDA) regulations, and national and international guidance documents that provide direction and lay out the framework for successfully implementing, maintaining, and sustaining an effective and robust quality management system. The standards, regulations, and guidances require the management of nonconformances and deviations for products and services provided. Risk-based thinking can help prioritize nonconformance and deviation management. The applicable standards, regulations, and guidances include, but are not limited to, the following:
ISO 9001:2015 — Quality management systems — Requirements
8.7.1 The organization shall ensure that outputs that do not conform to their requirements are identified and controlled to prevent their unintended use or delivery.
The organization shall take appropriate action based on the nature of the nonconformity and its effect on the conformity of products and services. This shall also apply to nonconforming products and services detected after delivery of products, during or after the provision of services.
ISO 13485:2016 — Medical devices — Quality management systems — Requirements for regulatory purposes
8.3.1 General -- The organization shall ensure that product which does not conform to product requirements is identified and controlled to prevent its unintended use or delivery. The organization shall document a procedure to define the controls and related responsibilities and authorities for the identification, documentation, segregation, evaluation and disposition of nonconforming product.
The evaluation of nonconformity shall include a determination of the need for an investigation and notification of any external party responsible for the nonconformity.
21 CFR 211 — Current Good Manufacturing Practice Finished Pharmaceuticals
Sec. 211.100 Written procedures; deviations.
(b) Written production and process control procedures shall be followed in the execution of the various production and process control functions and shall be documented at the time of performance. Any deviation from the written procedures shall be recorded and justified.
21 CFR 820 — Quality System Regulation
820.90 Nonconforming product.
(a) Control of nonconforming product. Each manufacturer shall establish and maintain procedures to control product that does not conform to specified requirements. The procedures shall address the identification, documentation, evaluation, segregation, and disposition of nonconforming product. The evaluation of nonconformance shall include a determination of the need for an investigation and notification of the persons or organizations responsible for the nonconformance. The evaluation and any investigation shall be documented.
(b) Nonconformity review and disposition.
(1) Each manufacturer shall establish and maintain procedures that define the responsibility for review and the authority for the disposition of nonconforming product. The procedures shall set forth the review and disposition process. Disposition of nonconforming product shall be documented. Documentation shall include the justification for use of nonconforming product and the signature of the individual(s) authorizing the use.
GHTF.SG3.N99-8 Guidance on Quality Systems for the Design and Manufacture of Medical Devices
4.13.1 General
When any intermediate or final product (including service) is found (e.g., by test or inspection) not to conform to the technical specifications, inadvertent use or installation should be prevented. This is applicable to nonconforming product occurring in the supplier's own production as well as nonconforming product received by the supplier.
An important element in addressing nonconformities is to give to all appropriate personnel the freedom to identify nonconforming items, activities and processes and encouragement to suggest improvements.
ICH Harmonized Tripartite Guideline Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients Q7
2.1 Principles
2.16 Any deviation from established procedures should be documented and explained. Critical deviations should be investigated, and the investigation and its conclusions should be documented.
ICH Harmonized Tripartite Guideline Pharmaceutical Quality System Q10
3.2.1 Process Performance and Product Quality Monitoring System
(e) Include feedback on product quality from both internal and external sources, e.g., complaints, product rejections, nonconformances, recalls, deviations, audits, and regulatory inspections and findings.
The above regulations, standards, and guidance documents either refer directly or indirectly to the use of risk-based thinking to manage nonconformances/deviations.
Get Mark's techniques for establishing and justifying the number of process validation runs:
How To Establish The Number of Runs Required For Process Validation
Nonconformance And Deviation Classification
Risk-based thinking should be an integral part of an effective and efficient nonconformance and deviation management program. The level of control should be proportionate to the effect on the quality of the product produced or services provided by your organization. It should be obvious that as the risk level of the nonconformance and deviation increases, so should the requirements and controls used to manage nonconformances and deviations.
Table 1 provides example definitions for low-, medium-, and high-impact nonconformances and deviations. Once the risk level has been determined (low, medium, or high), the appropriate risk-based nonconformance and deviation controls can be applied.
Table 1: Example Impact Definitions, Risk Acceptability, And Control Requirements
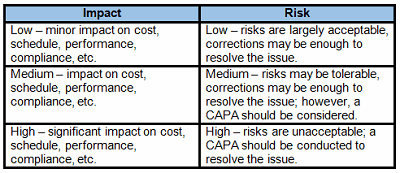
Another consideration for determining the impact and risk of nonconformances and deviations is repeat or recurring issues. Using Trending As A Tool For Risk-Based Thinking, an article published in September 2017, provides some additional guidance for the use of trending to identify and manage quality issues.
Nonconformance And Deviation Management
There are generally two methods to manage nonconformances and deviations. The first is through the nonconformances and deviations process; the second is the corrective and preventive action (CAPA) process. The CAPA process is primarily used for high- and medium-risk issues, while the nonconformances and deviations process is used for medium- and low-risk issues.
Table 2: Typical Corrective And Preventive Action Process Steps

The CAPA process has eight distinct steps or phases, including problem identification, impact assessment, remedial action/containment, investigation/root cause analysis, corrective action, implementation, verification of effectiveness, and closure. Each step has specific requirements that should be followed to ensure successful resolution of quality issues, including:
- Problem identification – describe the problem and its source
- Impact assessment – what products, processes, or systems may be affected
- Remedial action/containment – place product on hold, recall, quarantine, etc.
- Investigation/root cause analysis – determine what caused the issue
- Corrective action – actions taken to address the root cause of the problem
- Implementation – the deployment of corrective action(s)
- Verification of effectiveness – the plan, criteria, and requirements to ensure the problem will not recur or have other adverse effects.
- Closure – ensuring the corrective action(s) were effective; this should include a disposition record of any products or materials affected.
Table 3: Typical Nonconformance And Deviation Process Steps

The nonconformances and deviations process has six steps or phases, including problem identification, impact assessment, remedial action/containment, investigation/root cause analysis, correction, and closure. Each step has distinct requirements that should be followed to ensure successful resolution of quality issues, including:
- Problem identification – describe the problem and its source
- Impact assessment – what products, processes, or systems may be affected
- Remedial action/containment – place product on hold, recall, quarantine, etc.
- Investigation/root cause analysis – determine what caused the issue
- Correction – actions taken to address the root cause of the problem
- Closure – ensuring the correction was completed; this should include a disposition record of any products or materials affected
The CAPA process and the nonconformances and deviations process are very similar, except for corrective action vs. correction, implementation, and verification of effectiveness. To better understand these differences, Govind Ramu defines the difference as: “Correction is an action taken to eliminate a detected nonconformity,” and “Corrective action is taken to eliminate the cause of a detected nonconformity.” Ramu further states, “Both correction and corrective action may be required in many scenarios. Correction addresses the short-term need and gets immediate attention, and most organizations do a good job of correcting the nonconformity. Corrective action, on the other hand, is a long-term solution … organizations do not invest adequate resources in addressing corrective action.” Due to the short-term nature of corrections, the implementation and verification of effectiveness phases are generally not required or completed by most organizations.
Conclusion
The discussion above shows various opportunities for integrating risk management concepts to manage nonconformances and deviations. The concepts presented can be readily applied to virtually any industry as best practices.
The definitions and requirements presented in this article can and should be utilized based upon an organization’s risk acceptance threshold, industry practice, guidance documents, and regulatory requirements.
The methods presented here have been used and successfully defended during audits and inspections. I cannot emphasize enough the importance of documenting the methods and rationales your organization may use for managing risk activities.
This series of articles has introduced other methods for integrating risk management in the quality management system. The articles in the series include:
- Integrating Risk Management In The Quality Management System — A Primer
- An Introduction To qFMEA – A Tool For QMS Risk Management
- Using Risk-Based Thinking To Manage Suppliers
- Using Trending As A Tool For Risk-Based Thinking
- Contingency Plans: An Essential Quality Management System Risk Tool
References:
- Durivage, M.A., 2014, Practical Engineering, Process, and Reliability Statistics, Milwaukee, ASQ Quality Press
- Durivage, M.A., March 2017, Work Smarter, Not Harder: Make your CAPA verification of effectiveness SMART, Milwaukee, ASQ Quality Progress
- Ramu, Govind, August 2013, Expert Answers: Correction vs. Corrective Action, Milwaukee, ASQ Quality Progress
About The Author:
 Mark Allen Durivage is the managing principal consultant at Quality Systems Compliance LLC and an author of several quality-related books. He earned a B.A.S. in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. Durivage is an ASQ Fellow and holds several ASQ certifications, including CQM/OE, CRE, CQE, CQA, CHA, CBA, CPGP, CSQP, and CSSBB. He also is a Certified Tissue Bank Specialist (CTBS) and holds a Global Regulatory Affairs Certification (RAC). Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments.
Mark Allen Durivage is the managing principal consultant at Quality Systems Compliance LLC and an author of several quality-related books. He earned a B.A.S. in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. Durivage is an ASQ Fellow and holds several ASQ certifications, including CQM/OE, CRE, CQE, CQA, CHA, CBA, CPGP, CSQP, and CSSBB. He also is a Certified Tissue Bank Specialist (CTBS) and holds a Global Regulatory Affairs Certification (RAC). Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments.