
What Did ICH Q14 Miss On Analytical Method Validation?
By Joachim Ermer, Ph.D., GMP Compliance Adviser

The accuracy and reliability of analytical results are essential prerequisites for ensuring the quality, safety, and efficacy of medicinal products. Test methods used in the GMP environment must therefore be suitable, i.e., validated, for the respective application. This suitability must be proven before initial use and must also be ensured for each use over the entire life cycle.
The regulatory requirements for the analytical validation of test methods are described in ICH Guideline Q2(R2). Important key points for validation result from the method development described in ICH Q14, e.g., the analytical control strategy, robustness, or the use of development data in the validation strategy. Both guidelines should be considered together.
Goal Of Validation
The goal of validation is to determine and confirm the performance parameters of a test method in assessing whether it can guarantee the intended application correctly and reliably. Consequently, a well-founded validation is in the user's own interest. However, the prerequisite is the identification of performance parameters relevant for routine analysis and the user awareness of the requirements. Only then is a rational and effective check possible. Reliable knowledge of the performance parameters can also be used to optimize the effort involved in routine analysis, e.g., reducing the number of repeat analyses of samples and standards with sufficient precision.
It is important to ensure the performance of the test procedure over the entire life cycle, i.e., for every use. The initial validation prior to routine use should therefore only be regarded as one phase of the life cycle.
Regulatory Requirements
Proof of the suitability of analytical test methods is a general GMP requirement. While the EU GMP Guideline Part I generally requires "validated test methods," Part II refers to the ICH guidelines regarding the content of method validation. 21 CFR Part 211 also requires in §194(a)(2) that the suitability of the test methods must be verified (see Figure 1).
EU GMP Guide Part I
Chapter 6 Quality control:
6.15 Testing methods should be validated. [...]
EU GMP Guide Part II
Chapter 12 Validation:
12.81 Methods should be validated to include consideration of characteristics included within the ICH guidelines on validation of analytical methods. The degree of analytical validation performed should reflect the purpose of the analysis and the stage of the API production process.
21 CFR 211
§211.194 (a) (2):
[...] The suitability of all test methods used shall be verified under actual conditions of use.
Figure 1: Regulatory requirements for method validation
The ICH Guideline Q2A on validation of analytical procedures, published in 1994, was very important for the standardization of terms and definitions as well as for establishing the basic requirements for analytical method validation. The guideline was adopted by the EU in 1995 as a scientific guideline in the Notice to Applicants (EudraLex Volume 3) and is therefore binding for new authorizations. In 2005, the guidelines ICH Q2A and Q2B were merged under the name Q2(R1) with the new title Validation of analytical procedures: Text and Methodology, without changes to their content.
Since the 1990s, significant developments have occurred, particularly in pharmaceutical production, such as quality by design tools, risk analysis, and life cycle. The ICH validation guideline Q2(R1) focuses mainly on chromatographic test methods. Due to the lack of acceptance criteria, the basis to prove the required suitability is diffuse, and the chapter on "Linearity" confuses the response function (calibration model) with the linearity of the analyte in the sample, i.e., with the precision. The latter has led to problems with analytical techniques, which display nonlinear response functions, especially with biological methods and products.
In November 2018, the ICH therefore published a concept paper on the revision of the validation guideline and the introduction of a new guideline Q14 on analytical procedure development. The finalized guidelines in Step 4 were published on the ICH website on December 20, 2023 (valid since June 14, 2024).
Some of the shortcomings were rectified, but as expected, the international coordination process also led to various compromises and inconsistencies. The main changes (from the author's point of view) are shown in Figure 2.
Improvements, additions, & clarifications |
Ambiguities, lack of information, lack of context, errors |
|
Extension of the scope of application:
|
Life cycle aspects
|
|
Basis of demonstrating suitability:
|
Analytical target profile (ATP) is not mentioned (only in Q14) |
|
Use of existing knowledge:
|
No description of how to derive acceptance criteria (neither in text nor in examples |
|
Product-related range (reportable range):
|
Conflation of the validation study design with the final result (reportable result) |
|
Extensions regarding techniques:
|
|
|
Specificity/selectivity
|
|
|
"Linearity" replaced by "Response (calibration model)"
|
Response and calibration model are not necessarily identical
Nonlinear response:
Area% method:
|
|
Lower range limit
|
|
|
Accuracy:
|
|
|
Precision
|
The precision levels for the system, the measurement, the injection and precision of the final result are missing Aim of the precision studies is not discussed:
|
|
Consideration of uncertainty:
|
|
|
Combined assessment of accuracy and precision
|
|
| Illustrative examples for more detailed orientation |
|
Figure 2: Summary of improvements and shortcomings of ICH Q2(R2)
Life Cycle Of Analytical Test Methods
The classification of the (dossier) validation as part of the analytical life cycle was an important starting point in the 2018 ICH concept paper. Unfortunately, this aspect fell victim to the (extremely unfortunate) decision to pursue separate guidelines Q2 and Q14. Despite having its own chapter, the "life cycle" is extremely rudimentary and mentions the concept of revalidation, co-validation, and transfer only very generally. The core of the life cycle concept, i.e., ensuring performance over the entire life cycle, is completely ignored in ICH Q2(R2), and ongoing monitoring is not even mentioned in the glossary.
Despite the somewhat misleading title, the life cycle is well described in the new ICH Guideline Q14 Analytical Method Development from June 2024. In addition to the titular subject of method development, Q14 also addresses changes during the life cycle and the continuous monitoring of analytical performance. However, since Q2 is "interposed," it is more appropriate to speak of a "split" life cycle. A truly holistic life cycle concept is described in the general information chapter <1220> of the USP1.
The analytical life cycle begins with a clear objective, which must guide the upholding of the required suitability. According to ICH Q8, quality by design (QbD) is defined as a systematic approach that starts with a predefined goal and includes scientifically based and risk-controlled process understanding and control. Consequently, such a QbD approach should also be applied in quality control analytics.
The "predefined target", i.e., the requirements for the analysis, is represented by the concept of the analytical target profile (ATP),2 analogous to the quality target product profile (QTPP) in the manufacturing process. The performance requirements for the measurement of the respective quality attribute (e.g., content of active substance) are defined here. These are reflected by the required specification limits and refer to the reportable result, which is checked against the acceptance limits of the specification (see USP, General Notices). The paradigm shift is that the requirements for the final result are independent of the specific analytical test method. Any method that fulfills these requirements is in principle suitable for checking the respective quality attribute. This means that the ATP can serve as a reference point for the entire life cycle of the quality attribute and thus also for adjustments and changes in the life cycle of a specific test method. With appropriate regulatory acceptance, the continuous improvement process would be much easier to implement, as changes could be handled within the company quality system. Initial approaches to such regulatory flexibility are recognizable, e.g., in the examples in Q14 or as "established conditions" in ICH Q12.
The ATP summarizes the general performance requirements for the final result of the required measurement and may take the following form for a quantification test:
The test method must be able to quantify the active substance X in the presence of Y1, Y2, ...:
- over the range from A% to B% of the target concentration in the dosage form,
- with a precision of less than C% RSD,
- and an accuracy of less than D% error.

Alternative (corresponds to a combined assessment):
- so that 95% of the future final results are within ± w% of the true value.
All other performance parameters are either included in the required precision and accuracy (e.g., specificity) and/or method-specific and are identified after selecting a specific method, such as the calibration model.
In contrast to ICH Q14, there is no mention of ATP in the glossary for ICH Q2(R2).
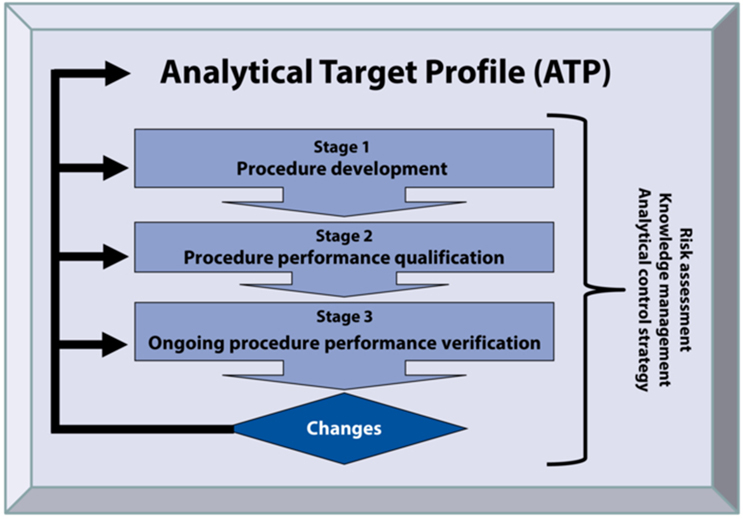
Figure 3: ATP and three-stage concept of the analytical procedure life cycle
In analogy to the validation of manufacturing processes according to the FDA’s Guideline on Process Validation, the general information chapter of the USP <1220> also defines three stages for the analytical procedure life cycle (see Figure 3).
In stage 1, the test procedure is developed and optimized (an "enhanced approach" according to ICH Q14).
Stage 2 confirms that the procedure meets the ATP requirements in the intended routine environment. This corresponds to traditional validation. However, the initially defined requirements of the ATP allow an iterative approach and the use of results from stage 1 in the final assessment, as described in ICH Q2(R2) and ICH Q14. In stage 2, the replication strategy of the final result is defined or confirmed on the basis of the precision studies, as well as the finalization of the analytical control strategy (e.g., system suitability test and sample acceptance criteria). Furthermore, the analytical method transfer and the implementation of pharmacopoeia procedures are also included in stage 2, as this also involves demonstrating the suitability of test procedures in the laboratory of use. If both laboratories are experimentally involved in the analytical transfer, the design corresponds to a comparative precision study with acceptance criteria for precision and accuracy (comparison of mean values). The performance parameters (USP <1226>) and acceptance criteria relevant for the verification of pharmacopoeia methods naturally correspond to those of validation and are to be defined depending on the application (i.e., monograph).
Stage 3 includes two aspects:
- ongoing monitoring, and
- performance review after changes.
Ensuring suitability for each application requires continual review by monitoring of performance parameters to identify and counteract undesirable trends at an early stage (FDA guidance on procedures and methods validation).3 Monitoring is mentioned in ICH Q14 (in contrast to ICH Q2(R2), but it includes no details. This gives the user the flexibility to identify suitable parameters. Performance parameters that can be calculated from normal routine analytical data, e.g., injection precision and span or RSD of sample preparations,4 are particularly suitable for this purpose. With additional analysis of control samples, even the long-term precision of the final result can be monitored using the same replication strategy as for the normal sample. In addition to the calculation of long-term and therefore very reliable averaged parameters (e.g., precision), control charts should be used to identify trends at an early stage. Like the regulator-required annual product review, the evaluation of the test procedure can be carried out regularly. An evaluation prior to the product review is recommended so that the conclusions can be adopted. Such an analytical evaluation — based on facts — reveals problems with the test procedures and allows the prioritization of continuous improvement activities.
After adjustments or changes to a procedure, the performance must be reviewed, depending on the degree of change, i.e., risk based. This is discussed in detail in ICH Q14 and corresponds to traditional revalidation. For example, changes within the robustness scope can be checked via continuous monitoring or the existing SST. In addition, stage 2 or possibly stage 1 must be considered. If a change of test method is intended, it is necessary to go through stage 1 again.
This article is an excerpt from GMP knowledge contained in the online portal GMP Compliance Adviser, which provides in-depth information about GMP best practices and regulations with a focus on Europe but also referring to the U.S., Japan, and many more (PIC/S, ICH, WHO, etc.).
References:
- USP. General Information Chapter <1220> The analytical procedure lifecycle.
- Schweitzer, M.; Pohl, M.; Hanna-Brown, M.; Nethercote, P.; Borman, P.; Hansen, G.; Smith, K.; Larew, J.; Carolan, J.; Ermer, J.; et al. Implications and opportunities of applying QbD principles to analytical measurements, Pharm. Tech. 2010, 34 (2).
- FDA. Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics. 2015.
- Ermer, J.; Aguiar, D.; Boden, A.; Ding, B.; Obeng, D.; Rose, M.; Vokrot, J. Lifecycle management in pharmaceutical analysis: How to establish an efficient and relevant continued performance monitoring program. J Pharm Biomed Anal 2020, 181, 113051. DOI: 10.1016/j.jpba.2019.113051.
About The Author:
 Joachim Ermer, Ph.D., is a biochemist and principal at Ermer Quality Consulting in Bensheim, Germany. He offers consulting and training on topics related to pharmaceutical analytics and quality control, such as pharmacopoeia requirements, reference standards, analytical data integrity, life cycle management of analytical procedures, monitoring of relevant performance parameters, continuous improvement, OOS/OOT, validation, verification, transfer or practical statistics. He is a member of the APV Analytical and Quality Assurance Specialist Group, the USP Measurement and Data Quality Expert Committee, and the Chromatographic Separation Techniques Working Party of the European Pharmacopoeia.
Joachim Ermer, Ph.D., is a biochemist and principal at Ermer Quality Consulting in Bensheim, Germany. He offers consulting and training on topics related to pharmaceutical analytics and quality control, such as pharmacopoeia requirements, reference standards, analytical data integrity, life cycle management of analytical procedures, monitoring of relevant performance parameters, continuous improvement, OOS/OOT, validation, verification, transfer or practical statistics. He is a member of the APV Analytical and Quality Assurance Specialist Group, the USP Measurement and Data Quality Expert Committee, and the Chromatographic Separation Techniques Working Party of the European Pharmacopoeia.