
An Introduction To Process Analytical Technology
By John D. Orr, Ph.D., and George L. Reid, III, Ph.D.

Our industry is engaged in the discovery, development, and production of high-quality, safe, and efficacious medicines intended for reducing the suffering and improving the lives of our patients. Ensuring medicinal product quality is our responsibility, and the FDA has written that “quality cannot be tested into products; it should be built in by design.” Many of our medicines are structurally complex or are made via complex processes (which include the use of hazardous materials). So, end-product testing alone is an insufficient measure of product quality, from our patients’ perspectives.
The use of well-designed and robust processes and analytical testing strategies (which include analyses of incoming materials, in-process materials, isolated process intermediates, drug substances, and finished drug product) enables us to confidently ensure product quality. This is an important responsibility and represents a significant amount of work that needs to be clearly documented and communicated to ensure efficient and effective review by regulatory authorities prior to product approval.
Process Analytical Background
Regulatory agencies understand the tremendous amount of work associated with ensuring product quality and have made efforts to encourage manufacturers to achieve success in this regard. In 2004, the U.S. FDA published an aspirational guidance for industry on process analytical technology (PAT). Unfortunately, more than a decade later, the routine use of PAT in an integrated manner to design, measure, and control critical process quality and performance attributes through timely and appropriate measurements remains largely unrealized across the pharmaceutical industry. Nevertheless, the use of PAT to understand and control pharmaceutical manufacturing processes (as appropriate) to ensure final product quality remains a worthy goal. The “PAT umbrella” comprises a broad suite of tools, including analytical, chemical, physical, microbiological, mathematical, and risk analysis. PAT interfaces the process with the analytical instrument, and may include a feedback loop to modify the process conditions based upon real-time analysis. This real-time data may aid both process control and product quality. Alternatively, using the traditional approach, the process is sampled and the sample is transported to a lab for analysis. This results in a delay between process sampling and when the results are available.
The ongoing debate over the use of PAT versus traditional analytical tools and controls may be one of semantics. In practice, pharma designs, develops, and controls processes via the use of appropriate and effective online and offline tools. The approaches used to ensure process understanding and control at a smaller scale and in an earlier stage of development may vary considerably from those used for larger-scale processes and in a later stage of development (when additional process and robustness knowledge is obtained).
Pharmaceutical Processes And Analyses
To help understand the apparent pedestrian uptake in PAT, it is helpful to understand pharmaceutical processes and analyses. Many processes are performed in batches. With solid oral drug products, for example, excipients and the drug substance are blended until they are uniform. This is done to ensure, to the individual tablet, composition is consistent. In traditional approaches, blend samples are taken in various positions in the blender to determine if the composition is homogeneous throughout. The act of blend sampling itself, though, may sometimes cause segregation within the sample and result in analysis bias.
In a PAT approach, a spectrometer is attached to the blender and spectra are acquired in real time to monitor the mixing. Once the spectra do not change any further, the blend is considered homogeneous. The limitation with the PAT approach, though, is the assumption that the sample in contact with the PAT spectrometer is representative of the entire blender contents. In the traditional offline approach, one is essentially “taking several snapshots, at the same time,” from samples obtained from different locations in the blender. In the PAT approach, one is essentially “recording a movie,” but only from one spot in the blender, with the assumption that the observations made there are representative of the entire blender.
With a drug substance, for example, reactants A and B are combined to form product C. The process is monitored for the disappearance of reactants A and B, and the formation of product C and potential byproducts. In traditional approaches, samples are periodically taken from the reactor, quenched, if need be, and brought to the testing lab. The samples are often analyzed by chromatography, where the individual components are separated from each other (in a time-dependent fashion) and quantified individually (Figure 1). This approach is analogous to “taking several snapshots, over time.”
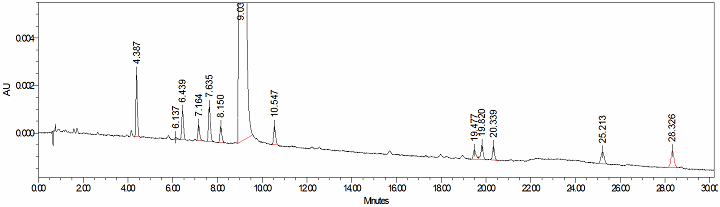
Figure 1: HPLC chromatogram. Each peak is an individual component, and quantitation is by the component peak area.
The limitations include fewer samples, longer analysis times, reactor perturbations from sampling, and the potential for the sample no longer being representative of the reactor mixture. The advantage is that the data analysis is more straightforward and the results are easy to visualize and interpret.
In a PAT approach, for example, the analysis technique is interfaced with the reactor for real-time analysis. Spectroscopic methods are often used, wherein a specific reactant, product, or byproduct molecular functional group characteristic (such as vibrational frequency or photon scatter frequency) can be monitored over time for disappearance (reactant) or appearance (product or byproduct). Method specificity is obtained via the specific frequencies selected (Figure 2). For example, if one is converting a carboxylic acid to an ester, one might monitor, by IR spectroscopy, the subtle shift in the carbonyl vibrational frequency (ca. 1700 cm-1) as the carboxylic acid is converted to an ester. Alternatively, other unique absorption frequencies may also be monitored.
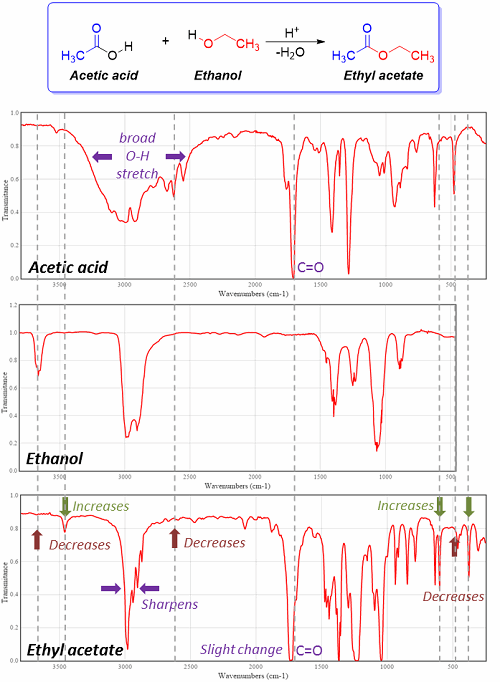
Figure 2: Reaction of acetic acid and ethanol, forming ethyl acetate, and the associated IR spectra of the individual components. The appearance, disappearance, or change of bands at representative wavenumbers allows for monitoring of a reaction.
The challenges of the PAT approach include the selection of the appropriate PAT technique and functional groups for reaction monitoring, spectral interferences resulting from components and process conditions, and the need for more sophisticated data analyses to visualize and interpret the results.
Barriers To Adoption Of PAT
The high degree of regulation of process, method, and controls differentiate the use of PAT in pharma as compared with other industries (petrochemical, chemical, and food). Pharmaceutical manufacturing is based on the use of cGMP controls to regulate the general operating systems and the application of validated manufacturing processes with well-defined operating ranges for each process for a marketed product. Once a process has been validated and approved, it is fixed until a change is accepted via a Change Being Effected notification (CBE) or a supplement requiring prior approval (PAS).
In many companies, especially those with limited resources, insufficient investment in the process development cycle creates a barrier to PAT. Resources are often restricted early in development (Phases 1 and 2). However, later in development (Phase 3), the time available to do process development work becomes a limitation. In such a paradigm, PAT implementation suffers.
In larger and more well-established pharmaceutical development organizations, there are analytical, process, and engineering groups. These groups all have different roles and deliverables, and often are within different organizational lines. In traditional product development, these lines interact, but largely function with a level of independence. The analyst will receive samples to evaluate, and may or may not have been involved in the experimental design and execution of the studies. The implementation of PAT requires changes to how processes are developed, analyzed, and produced. This necessitates an integrated co-development approach comprising the analysts, chemists, and engineers.

Photo courtesy of the National Institute for Bioprocessing Research & Training (NIBRT)
For technical staff, the selection of which technique to utilize, PAT or offline, is often not the key decision that must be made. Rather, the focus is on selecting the technique(s) that will provide the data (in the required amount of time with minimal resources) the project team needs to progress the project. Additional requirements include the scientists having the requisite knowledge, expertise, and instrumentation for the selected technique. During development, process control and analysis methods are defined, and the specific analysis needs for each process step (PAT or offline tools) are determined.
The final analysis and control methods are selected via collaboration between the development and manufacturing sites, company management, and regulatory affairs. To support (GMP) PAT activities, the receiving sites must have on-site technical experts, infrastructure (including explosion-proof PAT equipment and backups, ports for interfacing the probes, data analyzers, supervisory control and data acquisition [SCADA] systems), and appropriate quality systems. This results in a resource (human and financial)-intensive effort.
Analytical methods are not commonly critiqued during regulatory market application review. However, in the case of PAT, more scrutiny (for example, in areas of model development, maintenance, number of samples tested) is typical during regulatory market application review. This adds to a perceived increased regulatory burden, including country-specific requirements, more complex product life cycle management tasks, and increased time to product approval.
In the PAT initiative webpage of the FDA Office of Medicinal Products and Tobacco, the introduction states “The goal of PAT is to understand and control the manufacturing process, which is consistent with our current drug quality system: quality cannot be tested into products; it should be built-in or should be by design.” This is exactly right. Modern pharmaceutical processes are rationally developed using quality by design (QbD) principles.
Conclusions
Not all processes or products require the real-time multivariate monitoring and control that can be delivered using PAT. Most processes are sufficiently designed to operate without PAT. For those processes that would benefit from real-time monitoring, often the PAT measurement is correlated to a process parameter that can be routinely monitored with a low-technology (univariate) probe and sensor (e.g., pH, temperature, pressure, turbidity). Real-time process control is obtained, but with a much lower cost and resource requirement. During process risk assessments, high-risk areas are identified and appropriate controls are developed and implemented, some of which may include PAT measurements and feedback loops to control the process to produce a product with the target quality attributes. PAT can also be used when the act of sampling itself negatively impacts process safety, worker safety, or sample integrity. Additionally, some manufacturing processes (e.g., continuous) are well suited for the use of these real-time tools and feedback loops.
In this article, organizational construct, risk aversion, lack of (apparent) business drivers, resourcing the project, and perceived regulatory challenges were noted as impediments in adopting PAT routinely in pharma. In the next article in this three-part series, the authors discuss the application of spectroscopic measurements in lieu of more traditional analyses (e.g., chromatography, Karl Fischer) to provide familiarity with the tools, process, and typical samples. These spectroscopic tools, when integrated into a process, are termed PAT tools. In the final article, the authors discuss examples of where the PAT vision is realized, including the corporate/scientific attributes that led to this successful implementation.
Photo credit: Top/feature image courtesy of Sartorius
About The Authors:
 John D. Orr, Ph.D., managing advisor with Rondaxe Pharma, LLC, has 25 years of pharmaceutical chemistry, manufacturing, and controls (CMC) experience. He has been with Rondaxe since January 2016, assisting clients in the areas of CMC analytical and process development. Prior to joining Rondaxe, John served as director of analytical and physical chemistry at Eisai, where his group specialized in the pharmaceutical development of complex natural products.
John D. Orr, Ph.D., managing advisor with Rondaxe Pharma, LLC, has 25 years of pharmaceutical chemistry, manufacturing, and controls (CMC) experience. He has been with Rondaxe since January 2016, assisting clients in the areas of CMC analytical and process development. Prior to joining Rondaxe, John served as director of analytical and physical chemistry at Eisai, where his group specialized in the pharmaceutical development of complex natural products.
John has been active in several industry groups, serving on the board of directors of the IQ Consortium and the Allotrope Foundation. John received his chemistry degrees from the University of Pittsburgh (B.S.) and The University of Chicago (M.S. and Ph.D.), and performed post-doctoral studies in molecular biology at The Noble Foundation. You can reach him at jorr@rondaxepharma.com or connect with him via LinkedIn.
 George L. Reid, III, Ph.D., principal consultant with Cardinal Health Regulatory Sciences, has more than 20 years of pharmaceutical CMC experience, including regulatory, candidate progression from preclinical to post-approval, and technology development. He has contributed to several approved medicines and works with clients in the areas analytical, drug substance, and formulation development.
George L. Reid, III, Ph.D., principal consultant with Cardinal Health Regulatory Sciences, has more than 20 years of pharmaceutical CMC experience, including regulatory, candidate progression from preclinical to post-approval, and technology development. He has contributed to several approved medicines and works with clients in the areas analytical, drug substance, and formulation development.
George has been active in industry groups and currently is on the planning committee for the 2017 Land O'Lakes Pharmaceutical Analysis Conference. George earned doctorate in chemistry from Missouri University of Science and Technology and a B.S. in biochemistry from Beloit College. You can reach him at george.reid@cardinalhealth.com or connect with him via LinkedIn.